




THUỐC KHÁNG VIRUS ARN: HIV
Nếu nội dung bài viết chưa chính xác, vui lòng thông báo cho chúng tôi tại đây
Th9
THUỐC KHÁNG VIRUS ARN: HIV. Tải file PDF tại đây.
Cấu trúc và vòng đời của virus HIV
HIV (Hình 20.11) là một ví dụ về một nhóm virus được gọi là retrovirus. Có hai biến thể của HIV: HIV-1 gây ra bệnh AIDS ở Mỹ, Châu Âu và Châu Á, trong khi HIV-2 xảy ra chủ yếu ở Tây Phi. HIV đã được nghiên cứu rộng rãi trong 20 năm qua và nỗ lực nghiên cứu rộng lớn đã cho ra đời nhiều loại thuốc kháng virus đã được chứng minh là thành công trong việc làm chậm diễn tiến bệnh nhưng vẫn chưa có một loại thuốc nào điều trị được khỏi hoàn toàn bệnh. Hầu hết các loại thuốc kháng virus hữu ích trên lâm sàng hiện nay đều hoạt động dựa trên cơ chế: ức chế hai mục tiêu phân tử: enzyme phiên mã ngược và protease của virus. Do đó, cần phải phát triển các loại thuốc hiệu quả kháng lại mục tiêu phân tử thứ ba và những hiểu biết về vòng đời của HIV là điều kiện cần để xác định các mục tiêu phân tử phù hợp (Hình 20.11).
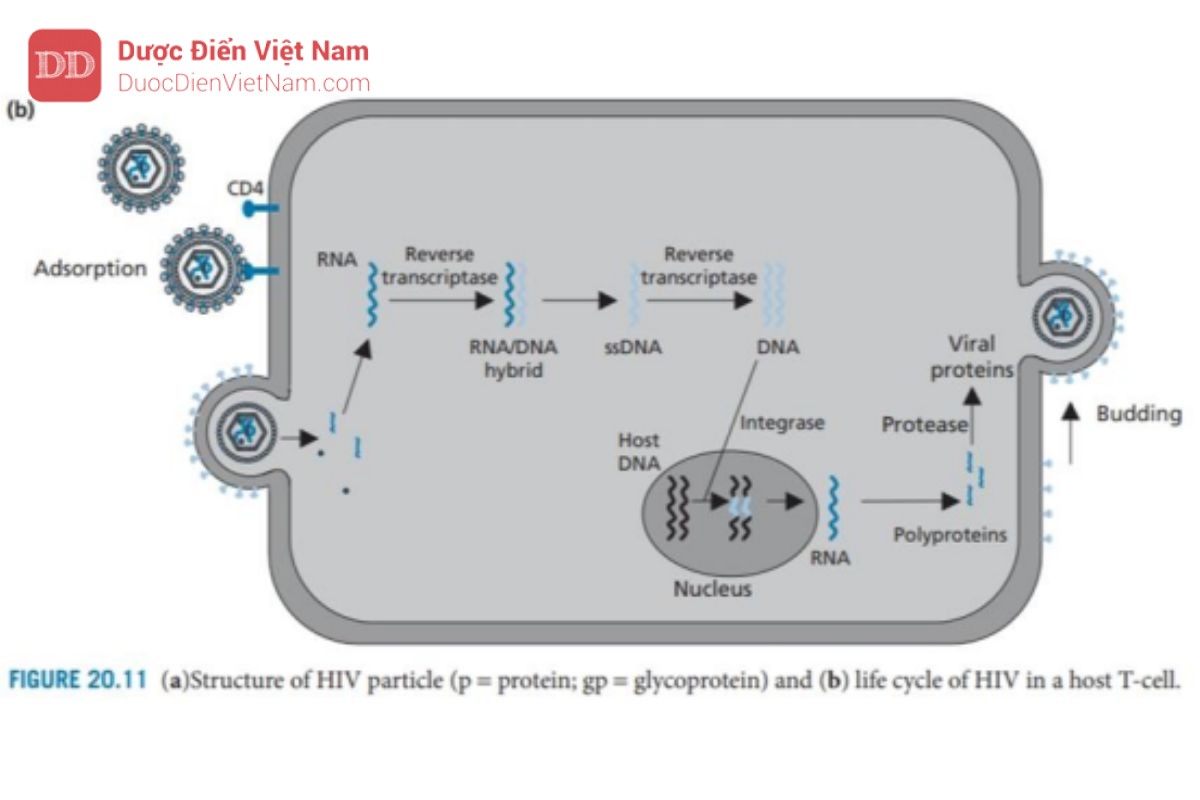
HIV là một loại virus RNA chứa hai chuỗi đơn RNA (+) giống hệt nhau trong lớp vỏ của nó. Ngoài ra, còn có các enzyme phiên mã ngược và integrase của virus, cũng như các protein khác gọi là p6 và p7. Capsid được tạo thành từ các đơn vị protein được gọi là p24; bao quanh vỏ capsid là một lớp ma trận protein (p17), sau đó là một lớp màng bao có nguồn gốc từ tế bào chủ và chứa các glycoprotein của virus gp120 và gp41.
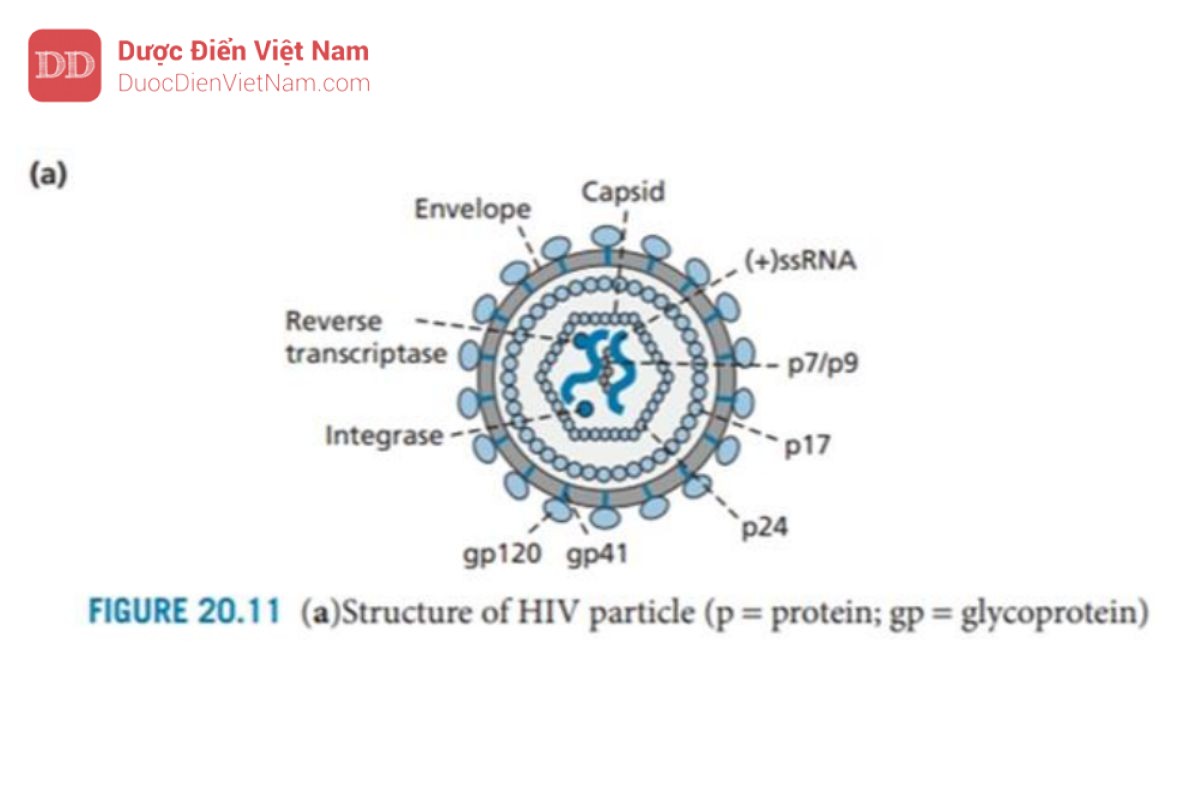
Cả hai loại protein này đều rất quan trọng đối với quá trình tiếp cận và xâm nhập. Gp41 đi qua lớp vỏ và liên kết cộng hóa trị với gp120 (phân tử này nhô ra khỏi bề mặt của virus).
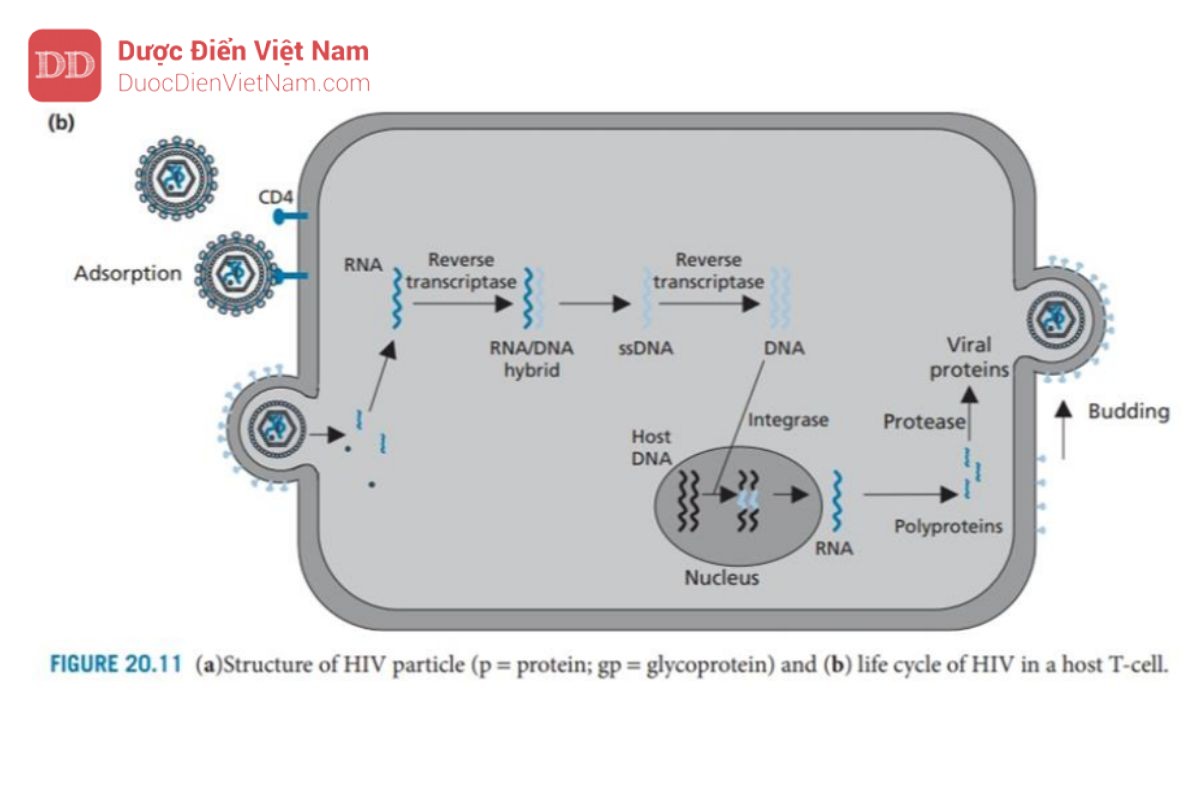
Khi virus tiếp cận tế bào chủ, gp120 tương tác và liên kết với protein xuyên màng CD4 có mặt trên tế bào lympho T của vật chủ. Sau đó, các protein gp120 thay đổi cấu hình cho phép chúng liên kết đồng thời với các thụ thể chemokine (CCR5 và CXCR4) trên tế bào chủ (không được hiển thị). Những thay đổi về hình dạng ở giai đoạn tiếp theo sẽ loại bỏ protein gp120 cho phép protein gp41 của virus tiếp cận bề mặt tế bào chủ và giữ virus trên bề mặt. Sau đó, gp41 thay đổi cấu hình giúp kéo virus và tế bào chủ lại với nhau để dung hợp màng.
Sau khi quá trình dung hợp đã diễn ra, nucleocapsid của HIV sẽ xâm nhập vào tế bào chủ. Sau đó, sự phân hủy protein Capsid diễn ra, có thể được hỗ trợ bởi hoạt động của một loại enzyme của virus có tên là protease. RNA virus và các enzyme virus sau đó được giải phóng vào tế bào chất của tế bào.
RNA virus được giải phóng không có khả năng mã hóa trực tiếp cho protein virus hoặc tự sao chép. Thay vào đó, nó được chuyển đổi thành DNA và tích hợp vào DNA của tế bào chủ. Việc chuyển đổi RNA thành DNA không phải là một quá trình xảy ra trong tế bào người, do đó không có enzyme của tế bào chủ xúc tác cho quá trình này. Do đó, HIV mang enzyme đặc trưng của nó – enzyme phiên mã ngược – để thực hiện việc này. Enzym này là thành viên của họ enzyme được gọi là DNA polymerase, nhưng điều bất thường là nó có thể sử dụng chuỗi RNA làm khuôn mẫu. Đầu tiên enzyme xúc tác quá trình tổng hợp chuỗi DNA sử dụng RNA virus làm khuôn tạo thành cấu trúc (+)RNA–(-)DNA. Enzyme phiên mã ngược xúc tác quá trình phân hủy chuỗi RNA sau đó sử dụng chuỗi đơn DNA còn lại làm khuôn để xúc tác quá trình tổng hợp chuỗi kép dsDNA (DNA tiền virus).
DNA tiền virus hiện được ghép vào DNA của tế bào chủ – một quá trình được xúc tác bởi enzyme tích hợp protein của virus. Khi DNA tiền virus đã được gắn vào DNA của vật chủ, nó được gọi là provirus và có thể tồn tại ở trạng thái không hoạt động trong DNA của tế bào chủ cho đến khi được kích hoạt bởi các quá trình tế bào.
Khi điều đó xảy ra, quá trình phiên mã của các gen virus env, gag và pol diễn ra để tạo ra RNA virut, một số trong đó sẽ được tích hợp vào các virion mới và phần còn lại được sử dụng trong quá trình dịch mã để tạo ra ba polyprotein lớn, không có chức năng, một loại có nguồn gốc từ gen env, một loại từ gen gag và loại còn lại từ gen gag-pol. Protein đầu tiên trong số này được phân cắt bởi các proteinase của tế bào và tạo ra các glycoprotein của virus (gp120 và gp41), được tích hợp vào màng tế bào chủ. Hai polypeptide còn lại (Pr55 và Pr160) vẫn còn nguyên vẹn và di chuyển tới mặt trong màng tế bào chủ. Các glycoprotein của virus trong màng tế bào cũng tập trung ở khu vực này và các protein của tế bào chủ bị loại trừ.
Sau đó quá trình nảy chồi diễn ra để tạo ra một hạt virus chưa trưởng thành, gắn trên màng. Trong quá trình nảy chồi, một enzyme của virus là protease được giải phóng khỏi polypeptide gag-pol. Điều này có được nhờ enzyme protease tự xúc tác sự phân cắt các liên kết peptide nhạy cảm liên kết nó với phần còn lại của polypeptide.
Sau khi được giải phóng, enzyme protease sẽ dimer hóa và cắt các chuỗi polypeptide còn lại để giải phóng enzyme phiên mã ngược, integrase và các protein cấu trúc của virus. Các protein capsid bây giờ tự lắp ráp để tạo thành các nucleocapsid mới chứa RNA virus, enzyme phiên mã ngược và integrase
Người ta đã quan sát thấy rằng một loại protein virus có tên là Vpu có vai trò quan trọng trong quá trình nảy chồi. Vpu liên kết với protein màng tế bào chủ CD4 và kích hoạt enzyme chủ để gắn protein CD4 với protein ubiquitin. Các protein được gắn ubiquitin sẽ được tế bào chủ đánh dấu để phá hủy và do đó các protein CD4 trong tế bào chủ sẽ bị loại bỏ. Điều này rất quan trọng vì các protein CD4 có thể tạo phức hợp với các protein virut mới được tổng hợp là gp120 và ngăn cản sự tập hợp của các virut mới.
Liệu pháp kháng virus chống lại HIV
Cho đến năm 1987, chưa có thuốc điều trị HIV, nhưng sự hiểu biết về vòng đời của HIV đã giúp xác định được một số mục tiêu phân tử tiềm năng. Hiện nay, hầu hết các loại thuốc đã được phát triển đều có tác dụng chống lại enzyme phiên mã ngược và protease của virus. Tuy nhiên, một vấn đề nghiêm trọng trong việc điều trị HIV là virus này dễ dàng biến đổi. Điều này dẫn đến tình trạng kháng thuốc nhanh chóng đối với thuốc kháng virus. Kinh nghiệm cho thấy nếu điều trị HIV bằng một loại thuốc đơn độc thì hiệu quả thu được chỉ ngắn hạn, nhưng về lâu dài, loại thuốc này chỉ có tác dụng chọn lọc những virus chưa có đột biến kháng thuốc. Kết quả là, liệu pháp điều trị hiện nay bao gồm sự kết hợp của nhiều loại thuốc khác nhau tác động lên cả enzyme phiên mã ngược và protease. Họ đã thành công trong việc trì hoãn sự tiến triển thành AIDS và tăng tỷ lệ sống sót, nhưng cần phải phát triển các loại thuốc hiệu quả để kháng lại mục tiêu phân tử thứ ba.
Nhu cầu đối với bất kỳ loại thuốc điều trị HIV nào cũng rất lớn, đặc biệt là vì nó có thể được sử dụng trong thời gian dài. Nó phải có ái lực cao với mục tiêu (trong khoảng nồng độ picomolar) và có hiệu quả trong việc ngăn chặn virus nhân lên và lây lan. Nó phải thể hiện ái lực kém đối với bất kỳ mục tiêu vật chủ tương tự trong tế bào, đồng thời phải an toàn và dung nạp tốt. Nó phải có tác dụng kháng
lại càng nhiều loại virus càng tốt, nếu không nó chỉ có tác dụng chọn lọc một vài biến thể. Nó cần phải có tác dụng hiệp đồng với các loại thuốc khác dùng để chống lại bệnh tật và tương thích với các loại thuốc khác dùng để điều trị các bệnh cơ hội và nhiễm trùng phát sinh do phản ứng miễn dịch suy yếu. Thuốc phải dung nạp ở trên ngưỡng nồng độ điều trị trong tế bào bị nhiễm bệnh và trong tuần hoàn. Nó phải có khả năng dùng bằng đường uống với tần suất dùng liều tối thiểu và tốt nhất là nó có khả năng vượt qua hàng rào máu não trong trường hợp virus ẩn náu trong não. Cuối cùng, nó phải không tốn kém vì nó có thể phải sử dụng suốt đời cho bệnh nhân.
BOX20
Không có cách chữa trị nhiễm HIV, nhưng thuốc chống HIV có thể ngăn chặn hoặc làm chậm tốc độ phát triển của bệnh, dẫn đến tăng đáng kể tuổi thọ. Thật không may, các loại thuốc được sử dụng đều có thể gây độc tính đặc biệt nghiêm trọng vì bệnh nhân phải dùng những loại thuốc này suốt đời. Điều này có nghĩa là bệnh nhân phải được theo dõi liên tục. Một sự kết hợp của các loại thuốc tác dụng kháng lại hai mục tiêu enzyme khác nhau được sử dụng— liệu pháp kháng vi-rút hoạt tính cao (HAART). Khi lựa chọn sử dụng loại thuốc nào, điều quan trọng là phải đảm bảo rằng chúng có tác dụng hiệp đồng hoặc tác dụng phụ và tương thích về độc tính hại.
Hiện tại, thuốc ức chế protease (PI) (mục 20.7.4) được sử dụng cùng với thuốc ức chế men sao chép ngược (mục 20.7.3) (liệu pháp phân kỳ) hoặc với một PI khác (liệu pháp hội tụ). Nên kết hợp hai thuốc ức chế men sao chép ngược nucleoside (NRTI) với một PI, nhưng người ta cũng có thể sử dụng hai PI với một NRTI, hoặc một chất ức chế men sao chép ngược không nucleoside (NNRTI) với hai NRTI. Ví dụ, NNRTI efavirenz được sử dụng cùng với NRTI emtricitabine và tenofovir.
Các NRTI có thể được sử dụng để chống lại HIV là zidovu dine, didanosine, zalcitabine, stavudine, lamivudine, emtricitabine, tenofovir disoproxil và abacavir. NNRTI được sử dụng để điều trị HIV là nevirapine, delavirdine và efavirenz. Các PI được sử dụng là saquinavir, ritonavir, indinavir, nelfi navir, amprenavir, atazanavir, darunavir, fosamprenavir, lopinavir và tipranavir
Chất ức chế phản ứng tổng hợp enfuvirtide hiện đã được phê duyệt là thuốc chống HIV và có tác dụng chống lại mục tiêu khác với các loại thuốc chống HIV thông thường. Nó có thể được dùng cùng với các thuốc thông thường nếu bệnh không đáp ứng với liệu pháp HAART tiêu chuẩn. Chất ức chế integrase đầu tiên cũng đã được phê duyệt để sử dụng trong lâm sàng.
Nhờ HAART, tỷ lệ tử vong do các ca tử vong liên quan đến AIDS dường như đang chậm lại. Ví dụ, 1,8 triệu ca tử vong được ghi nhận vào năm 2009 so với 2,4 triệu vào năm 2004.
Chất ức chế enzym phiên mã ngược của virus
Chất ức chế enzym phiên mã ngược nucleoside (NRTI)
Vì enzyme phiên mã là enzyme chỉ có ở virus HIV nên đây được xem là mục tiêu phân tử lý tưởng của thuốc. Tuy nhiên, enzyme này vẫn mang bản chất là DNA polymerase nên phải thận trọng khi nghiên cứu phát triển các chất ức chế để chúng không có tác dụng ức chế đáng kể đối với DNA polymerase của người. Nhiều cấu trúc khác nhau kiểu nucleoside tỏ ra hữu ích trong việc kháng virus. Phần lớn trong số này ở dạng tiền thuốc khi vào trong tế bào chúng được chuyển thành dạng nucleotide triphosphate có hoạt tính nhờ phản ứng phosphoryl hóa bởi ba enzyme của tế bào chủ. Đây là quá trình tương tự đã được mô tả trước đây trong phần 20.6.1, nhưng có một điểm khác biệt quan trọng: các enzyme của tế bào cần phải xúc tác cả ba quá trình phosphoryl hóa vì HIV không mang enzyme kinase.
Zidovudine (Hình 20.12) ban đầu được phát triển như một chất kháng ung thư nhưng là loại thuốc đầu tiên được chấp thuận sử dụng trong điều trị bệnh AIDS. Zidovudine có cấu trúc tương tự như một deoxythymidine trong đó bộ phận đường 3′-hydroxyl đã được thay thế bằng nhóm azido. Khi chuyển thành dạng triphosphate, nó ức chế enzyme phiên mã ngược. Hơn nữa, triphosphate được gắn vào chuỗi DNA khi đang kéo dài lúc này nhóm 3′-hydroxyl đã được thay thế bằng nhóm azido nên chuỗi axit nucleic không thể kéo dài thêm nữa quá trình phiên mã ngược dừng lại
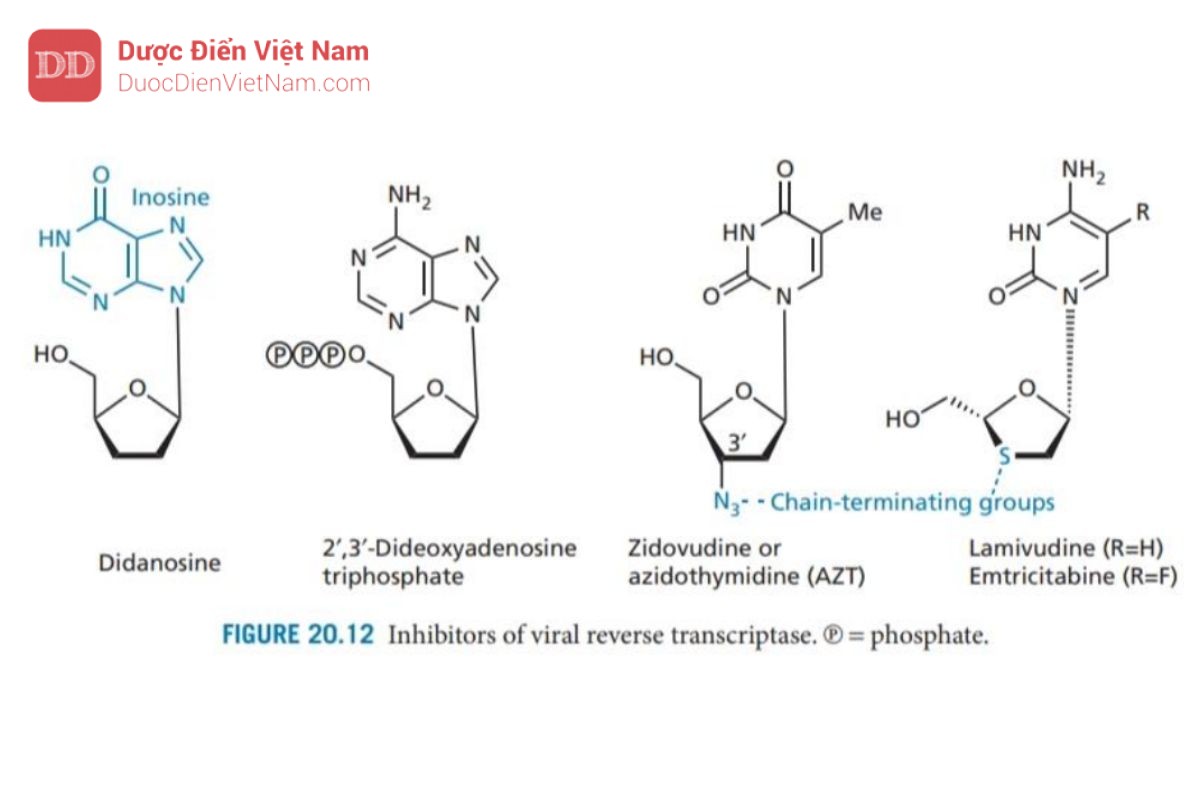
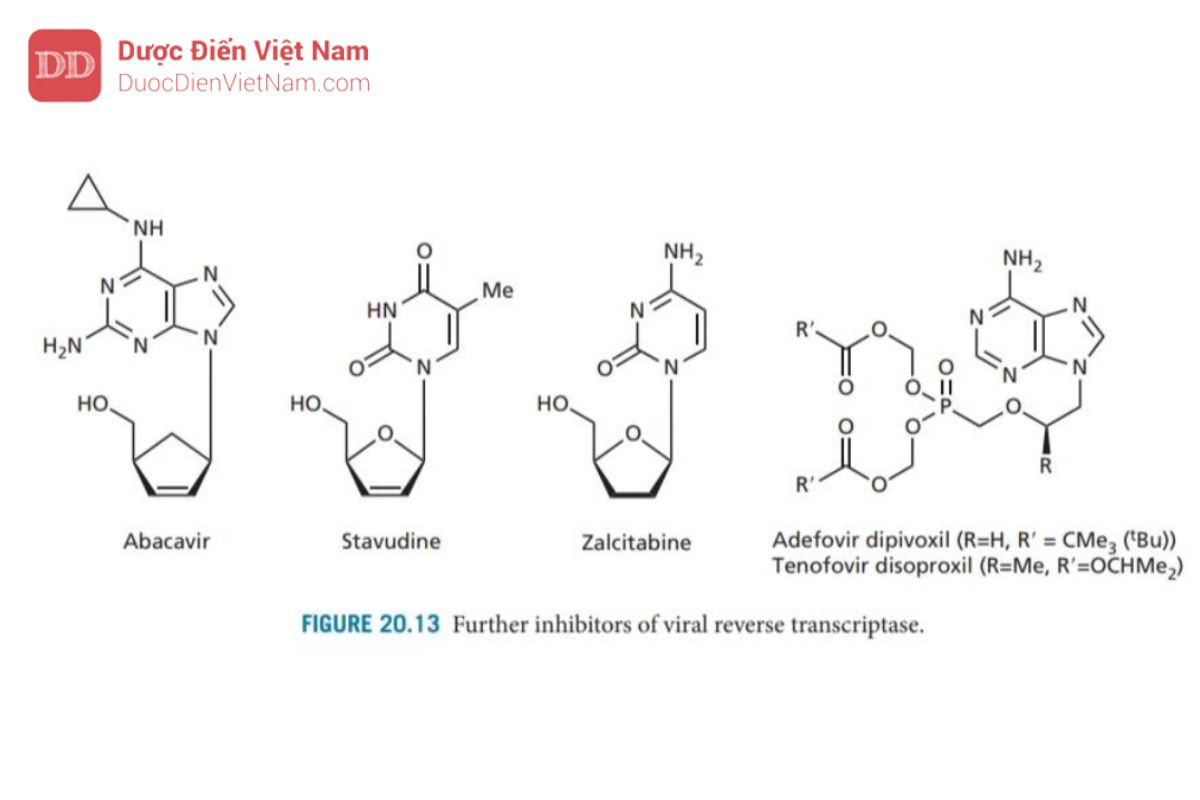
Didanosine (Hình 20.12) là loại thuốc kháng HIV thứ hai được chấp thuận sử dụng ở Hoa Kỳ (1988). Cơ chế tác dụng: Một base N của axit nucleic là inosine (một bazơ N không xuất hiện trong tự nhiên) được gắn vào đại phân tử DNA. Tuy không xuất hiện trong tự nhiên nhưng khi vào tế bào một loạt các phản ứng enzyme chuyển hóa hợp chất này thành 2′,3′-dideoxyadenosine triphosphate là thuốc có hoạt tính. Các nghiên cứu về trung tâm hoạt động của enzyme mục tiêu đã dẫn đến sự phát triển của lamivudine và emtricit abine (Hình 20.12) (cấu trúc tương tự như deoxycytidine trong đó carbon 3′ đã được thay thế bằng lưu huỳnh). Các NRTI hữu ích về mặt lâm sàng khác được sử dụng để kháng HIV và/hoặc viêm gan B bao gồm abacavir (chất có cấu trúc tương tự guanosine duy nhất), stavudine và zalcitabine (Hình 20.13). Tenofovir disoproxil và adefovir Dipivoxil là tiền chất của các nucleoside được biến đổi. Cả hai cấu trúc đều chứa một nhóm monophosphate được bảo vệ bởi hai este mở rộng. Quá trình thủy phân in vivo cho thấy nhóm photphat sau đó có thể được phosphoryl hóa thành triphosphate mang hoạt tính như đã mô tả trước đây.
Các chất ức chế enzym phiên mã ngược không có cấu trúc nucleoside (NNRTIs)
Nhìn chung các NNRTIs (Hình 20.14) là các phân tử kỵ nước liên kết với vị trí liên kết dị lập thể có bản chất kỵ nước trên enzyme phiên mã ngược. Vì vị trí liên kết dị lập thể tách biệt với vị trí liên kết (trung tâm hoạt động của enzyme phiên mã ngược) nên NNRTIs là chất ức chế không cạnh tranh và thuận nghịch. Chúng bao gồm các NNRTIs thế hệ thứ nhất, như nevirapine và delavirdine, cũng như các thuốc thế hệ thứ hai, như efavirenz, etravirine và rilpivirine. Các nghiên cứu về tinh thể của phức hợp enzyme-chất ức chế bằng phương pháp nhiễu xạ tia X đã chứng minh vị trí liên kết dị lập thể nằm liền kề với vị trí liên kết. Sự liên kết của NNRTIs với các vị trí dị lập thể dẫn đến sự thay đổi cấu trúc không gian tại vị trí liên kết trong trung tâm hoạt động của enzyme phiên mã ngược dẫn đến vị trí liên kết cơ chất bị khóa và trở thành một cấu hình không hoạt động. Thật đáng tiếc, tình trạng kháng thuốc nhanh chóng xuất hiện do đột biến ở vị trí liên kết với NNRTIs – phổ biến nhất là việc thay thế Lys 103 bằng asparagine. Đột biến này được gọi là K103N và được định nghĩa là đột biến kháng thuốc lớp Pan. Vấn đề kháng thuốc có thể được khắc phục bằng cách kết hợp NNRTIs với NRTI ngay từ khi bắt đầu điều trị. Hai loại thuốc này có thể được kết hợp sử dụng cùng nhau vì vị trí liên kết khác nhau.
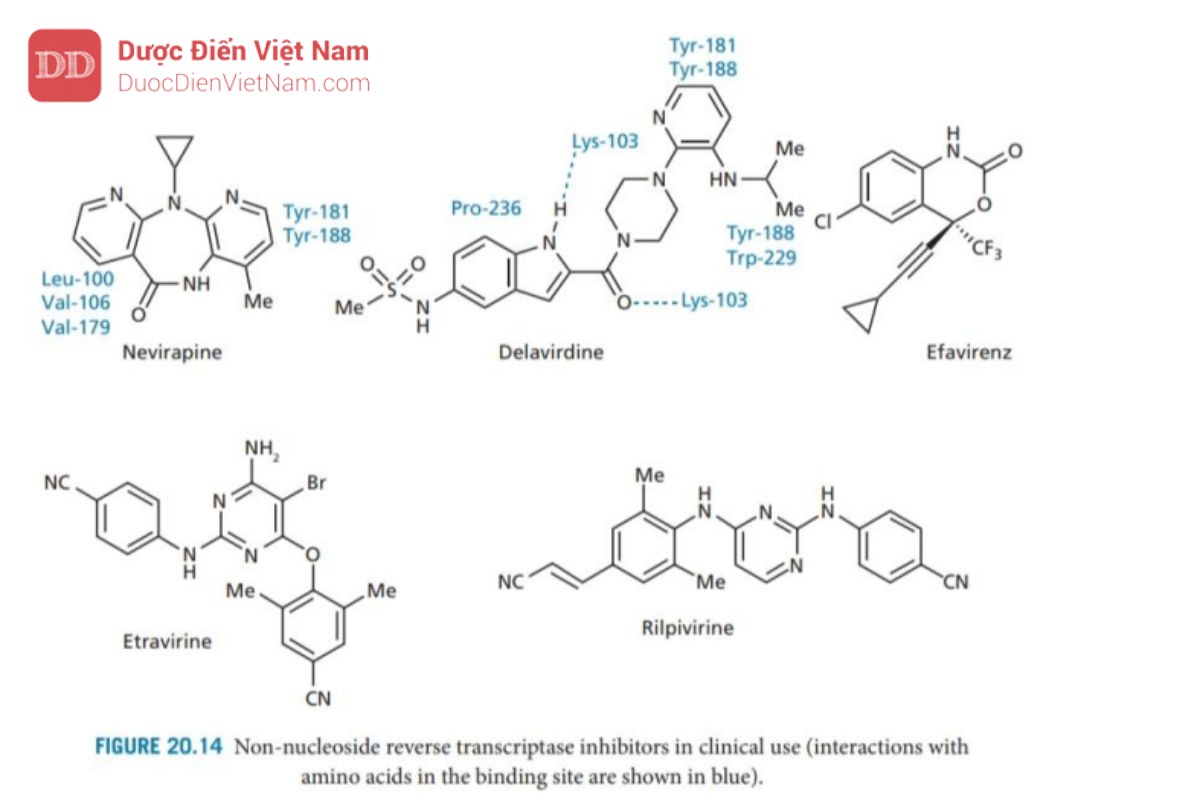
Nevirapine được phát triển từ một chất dẫn đường được phát hiện thông qua một chương trình sàng lọc ngẫu nhiên và có cấu trúc cứng nhắc giống như con bướm khiến nó có tính kháng khuẩn. Một ‘cánh’ tương tác thông qua tương tác kỵ nước và van der Waals với acid amin có nhân thơm (Tyr_181 và Tyr_188) ở vị trí liên kết, trong khi cánh còn lại tương tác với các acid amin no mạch hở (Leu_100, Val_106 và Val_179). Các chất ức chế NNRTIs khác liên kết với cùng một túi thân dầu và hoạt động như các chất cho điện tử π cho các nhân thơm trên của các acid amin.
Delavirdine được phát triển từ một hợp chất dẫn đường được phát hiện trong chương trình sàng lọc gồm 1500 hợp chất có cấu trúc đa dạng. Nó có kích thước lớn hơn các NNRTIs khác và một phần cấu trúc của nó nằm ngoài túi thân dầu bình thường để tham gia tương tác với dung môi xung quanh. Cấu trúc vòng pyridine và nhóm iso propylamine là những phần nằm sâu nhất trong vị trí liên kết tại trung tâm dị thể chúng tương tác với acid amin tyrosine và tryptophan. Ngoài ra còn cấu trúc này còn giúp mở rộng các tương tác kỵ nước. Không giống như các NNRTIs thế hệ đầu tiên khác, có liên kết hydro với aicd amin Lys-103 của các chuỗi peptide bên cạnh. Vòng indole của Delavirdine tương tác với Pro-236 và các đột biến liên quan đến Pro-236 dẫn đến kháng thuốc. Chất tương tự có vòng pyrrole thay cho indole có thể tránh kháng thuốc.
NNRTIs thế hệ hai được phát triển đặc biệt để tìm ra các tác nhân có hoạt tính chống lại các biến thể kháng thuốc, cũng như các loại virus hoang dã. Sự phát triển này đã được hỗ trợ bởi các nghiên cứu về tinh thể học bằng phương pháp nhiễu xạ tia X kết quả thu được cho biết cách thức các chất ức chế liên kết với vị trí liên kết trên trun tâm dị lập thể như thế nào. Từ các nghiên cứu giải trình tự đã chỉ ra rằng trong hầu hết các đột biến gây kháng NNRTIs thế hệ thứ nhất, một axit amin lớn được thay thế bằng một axit amin nhỏ hơn, dẫn đến sự tương tác thân dầu quan trọng bị mất. Điều thú vị là, các đột biến thay thế một axit amin bằng một axit amin lớn hơn có vẻ gây bất lợi cho hoạt động của enzyme, nhưng không tìm thấy đột biến nào ngăn cản NNRTIs xâm nhập vào vị trí liên kết. Efavirenz là một cấu trúc benzoxazinone có hoạt tính. chống lại nhiều biến thể đột biến nhưng có hoạt tính yếu hơn trên biến thể đột biến K103N. Tuy hoạt tính giảm so với nevirapine và kết quả nghiên cứu cấu trúc tinh thể bằng nhiễu xạ tia X của từng phức hợp cho thấy nhóm cyclopropyl của efavirenz có tương tác với Tyr-181 và Tyr-188 yếu hơn nevirapine nên những đột biến kháng thuốc của các axit amin này ít có hiệu quả trên efavirenz so với trên nevirapine. Efavirenz cũng là một cấu trúc nhỏ hơn và có thể thay đổi vị trí liên kết khi xảy ra đột biến K103N, cho phép nó hình thành liên kết hydro với chuỗi peptide của vị trí liên kết.
Các nghiên cứu cấu trúc tinh thể bằng nhiễu xạ tia X của các phức hợp enzyme với một số NNRTIs thế hệ hai cho thấy các tác nhân này chứa phân nửa cấu trúc không thơm tương tác với các gốc thơm Tyr-181, Tyr-188 và Trp 229 ở đầu túi liên kết. Khối lượng tương đối nhỏ và khả năng hình thành liên kết hydro với chuỗi peptide đóng vai trò quan trọng vì chúng cho phép các hợp chất thay đổi cách thức liên kết khi xảy ra đột biến. Các NNRTIs gần đây nhất được phê duyệt là etravirine (2008) và rilpivirine (2011).
Chất ức chế protease
Vào giữa những năm 1990, việc sử dụng phương pháp nhiễu xạ tia X để nghiên cứu cấu trúc tinh thể và mô hình phân tử đã giúp thiết kế cấu trúc của một loạt chất ức chế enzyme protease của virus HIV. Giống như các chất ức chế enzyme phiên mã ngược, chất ức chế protease (PIs) có hiệu quả trong thời gian ngắn khi sử dụng đơn độc do tình trạng kháng thuốc xuất hiện sớm. Do đó, liệu pháp phối hợp hiện nay là phương pháp được chấp nhận để điều trị nhiễm HIV. Khi sử dụng đồng thời các chất ức chế enzyme phiên mã ngược và protease, hoạt tính kháng virus sẽ được tăng cường và khả năng xuất hiện biến thể kháng thuốc sẽ chậm lại.
Không giống như các chất ức chế enzyme phiên mã ngược, PIs không phải là tiền thuốc và không cần phải hoạt hóa. Vì vậy, có thể sử dụng các thử nghiệm in vitro trên các tế bào bị nhiễm virus để đánh giá hoạt tính kháng virus của chúng. Enzym protease cũng có thể được phân lập, cho phép thực hiện các thử nghiệm trên enzyme này.
Phương pháp thứ hai để đánh giá mức độ hiệu quả của chất ức chế enzym protease là sử dụng IC50 như một. IC50 là nồng độ thuốc cần thiết để ức chế 50% enzyme. Như vậy, giá trị IC50 càng thấp thì tác dụng của chất ức chế càng mạnh. Tuy nhiên, PIs tốt không nhất thiết có nghĩa là thuốc kháng virus tốt. Để có hiệu quả, thuốc phải đi qua màng tế bào của tế bào bị nhiễm virus, do đó các thử nghiệm in vitro diễn ra trên toàn tế bào thường được sử dụng cùng với các nghiên cứu về enzyme để đánh giá sự hấp thụ của tế bào. Giá trị EC50 là thước đo hoạt tính kháng virus và thể hiện nồng độ các hợp chất cần thiết để ức chế 50% tác động gây bệnh tế bào của virus trong các tế bào lympho phân lập. Một điều phức tạp khác là yêu cầu thuốc chống HIV phải có sinh khả dụng tốt qua đường uống. Đây là một vấn đề đặc biệt với PIs. Như chúng ta sẽ thấy, hầu hết các PIs đều được thiết kế từ các hợp chất dẫn đường có cấu trúc peptit. Các peptit thường có trọng lượng phân tử cao, độ tan trong nước kém và các liên kết peptide nhạy cảm về chuyển hóa. Trong các ví dụ sau, chúng ta sẽ thấy rằng các PIs
mạnh được phát hiện tương đối nhanh, nhưng chúng có đặc tính peptide cao. Công việc tiếp theo là cần thiết để giảm đặc tính peptit của các hợp chất này nhằm duy trì hoạt tính tốt, đồng thời đạt được mức sinh khả dụng đường uống và thời gian bán hủy có thể chấp nhận được.
Các PIs hữu ích trên lâm sàng thường được hấp thu qua đường tiêu hóa kém hơn so với các chất ức chế enzyme phiên mã ngược và cũng dễ bị ảnh hưởng bởi các phản ứng chuyển hóa bước 1 qua đường tiêu hóa liên quan đến isozyme cytochrome P450 (CYP3A4). Sự trao đổi chất có thể dẫn đến tương tác thuốc-thuốc với nhiều loại thuốc khác được dùng cho bệnh nhân AIDS để chống lại các bệnh cơ hội (ví dụ rifabutin, ketoconazole, rifampin và astemizole).
Enzim protease HIV
Enzym protease HIV (Hình 20.15) là một ví dụ về họ enzyme được gọi là aspartyl protease – các enzyme xúc tác sự phân cắt các liên kết peptide và chứa axit aspartic ở vị trí hoạt động rất quan trọng đối với cơ chế xúc tác. Enzim này tương đối nhỏ và có thể thu được bằng cách tổng hợp. Ngoài ra, nó có thể được nhân bản và biểu hiện trong các tế bào phát triển nhanh sau đó được tinh chế với số lượng lớn. Enzim này được kết tinh có hoặc không có chất ức chế liên kết tại vị trí hoạt động, khiến nó trở thành mục tiêu phân tử lý tưởng cho việc thiết kế thuốc mới dựa trên cấu trúc tinh thể của phức hợp chất ức chế enzyme.
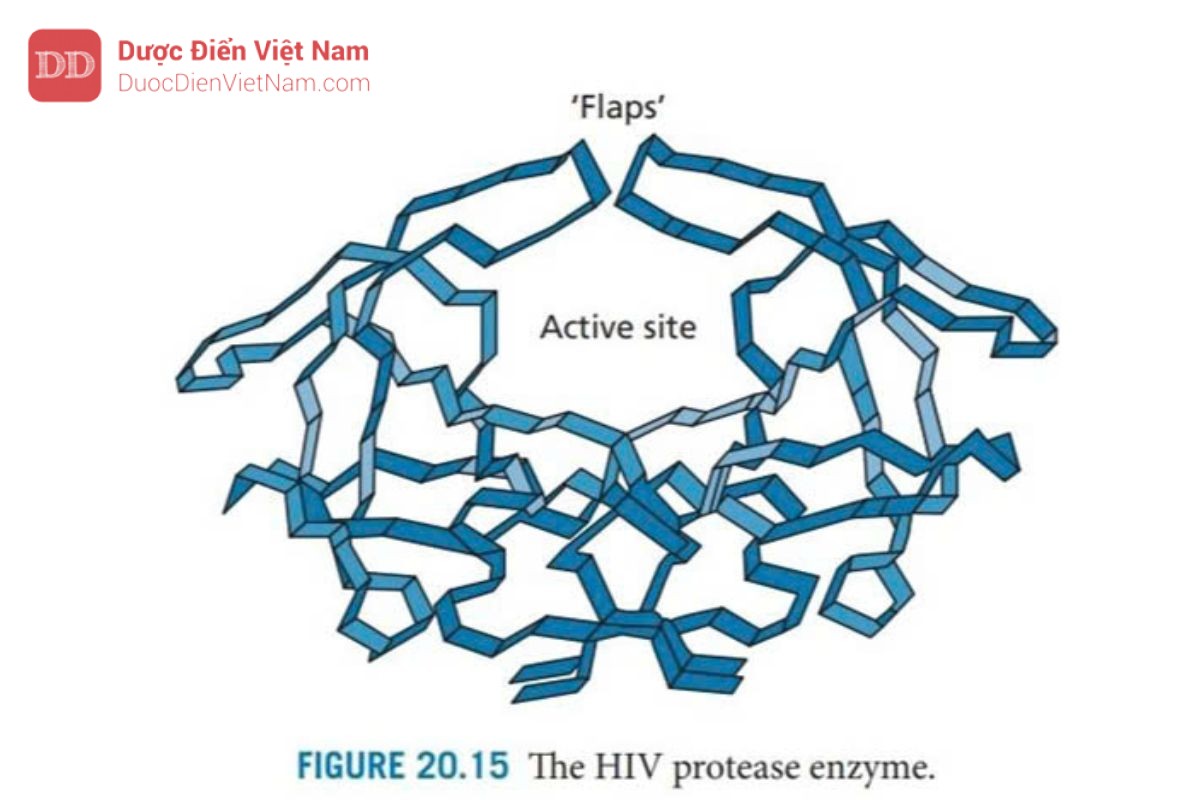
Enzym protease HIV là một dimer đối xứng được tạo thành từ hai tiểu đơn vị protein giống hệt nhau, mỗi đơn vị bao gồm 99 axit amin. Vị trí hoạt động nằm ở bề mặt phân cách giữa các đơn vị protein và cũng có tính đối xứng với sự đối xứng quay kép (C2). Các axit amin Asp-25, Thr-26 và Gly-27 từ mỗi monome nằm trên mặt phẳng của trung tâm hoạt động và mỗi monome cung cấp một nắp để hoạt động như mặt phẳng. Enzim này có phổ xúc tác rộng và có thể phân cắt nhiều loại liên kết peptit trong các polypeptide của virus, nhưng điều quan trọng là nó có thể phân cắt các liên kết peptid giữa acid amin proline và acid amin mang nhân thơm (phenylalanine hoặc tyrosine) (Hình 20.16). Sự phân tách liên kết peptide bên cạnh proline không xảy ra với các protease của động vật có vú như renin, pepsin hoặc cathepsin D, do đó các chất ức chế enzyme protease HIV có thể sẽ mang tính chọn lọc đủ tốt. Hơn nữa, tính đối xứng của trung tâm hoạt động của enzyme này không gặp ở các protease của động vật có vú, một lần nữa cho thấy khả năng chọn lọc của thuốc.
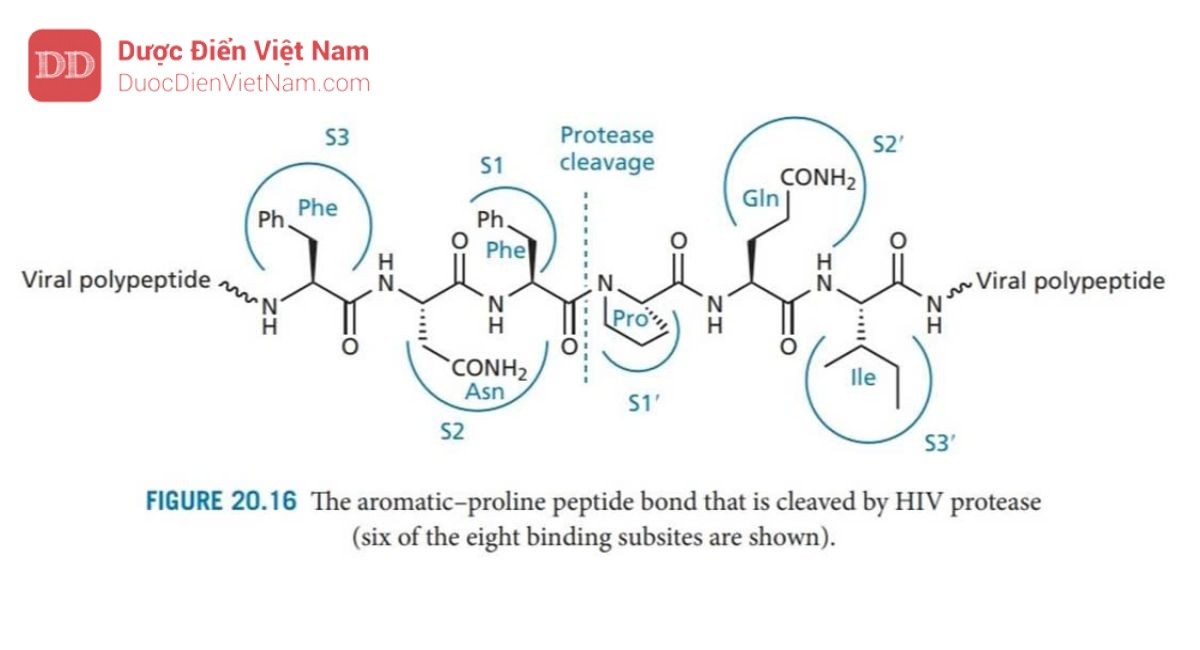
Có tám vị trí liên kết trong enzyme—bốn vị trí trên mỗi tiểu đơn vị protein, nằm ở hai bên của vùng xúc tác (Hình 20.16). Các vị trí này tiếp nhận các nhóm thế của axit amin trong phân tử cơ chất peptid và được đánh số S1–S4 ở tiểu đơn thứ nhất và S1′– S4′ ở tiểu đơn vị còn lại. Các nhóm thế tham gia liên kết với enzyme trên phân tử cơ chất được đánh số P1–P4 và P1′–P4′ (Hình 20.17). Các liên kết peptit trong cơ chất cũng tạo liên kết hydro với vị trí hoạt động, như trong Hình 20.17. Một phân tử nước có mặt ở trung tâm hoạt động, đóng vai trò là cầu nối liên kết hydro với hai nhóm NH isoleucine trên nắp enzyme. Mạng lưới liên kết hydro có tác dụng đóng các nắp trên vị trí hoạt động khi chất nền được liên kết.
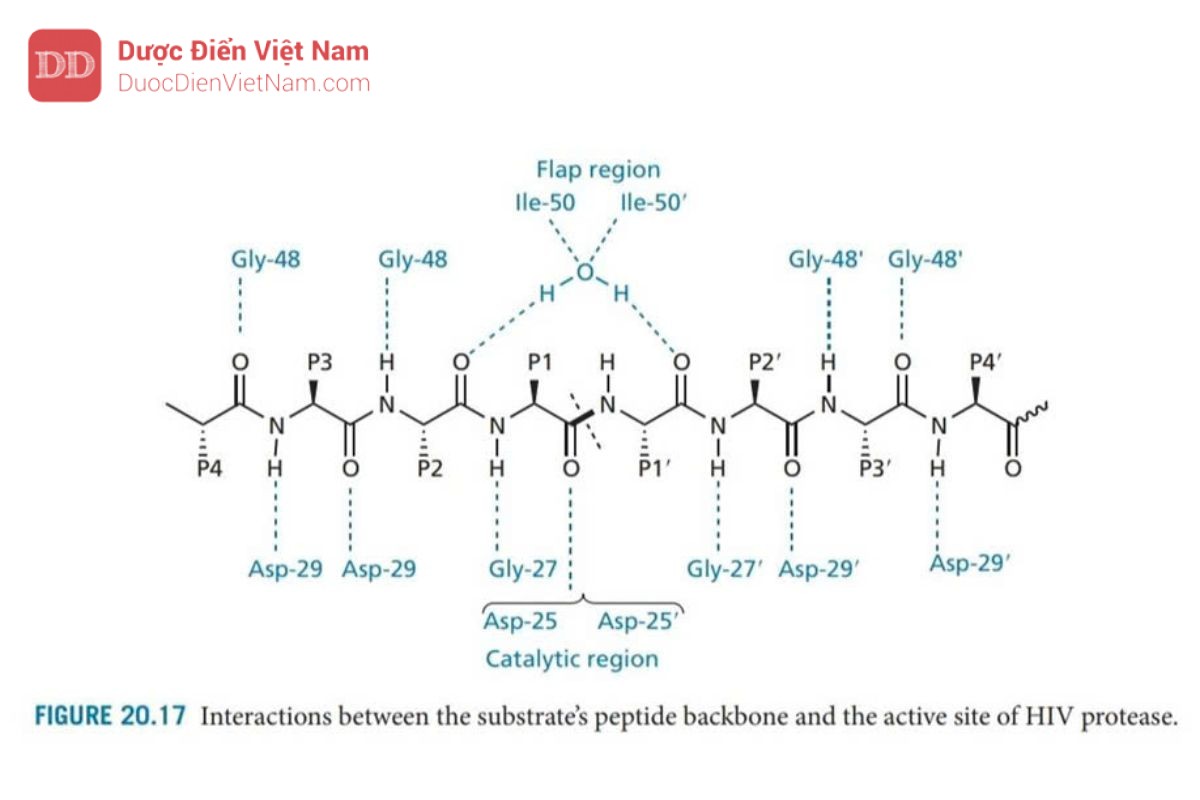
Có hai biến thể của HIV protease. Enzym protease của HIV-2 có 50% trình tự giống với enzyme protease của HIV-1. Sự biến đổi lớn nhất xảy ra bên ngoài vị trí hoạt động và do đó các chất ức chế được phát hiện là có khả năng liên kết với cả hai enzyme.
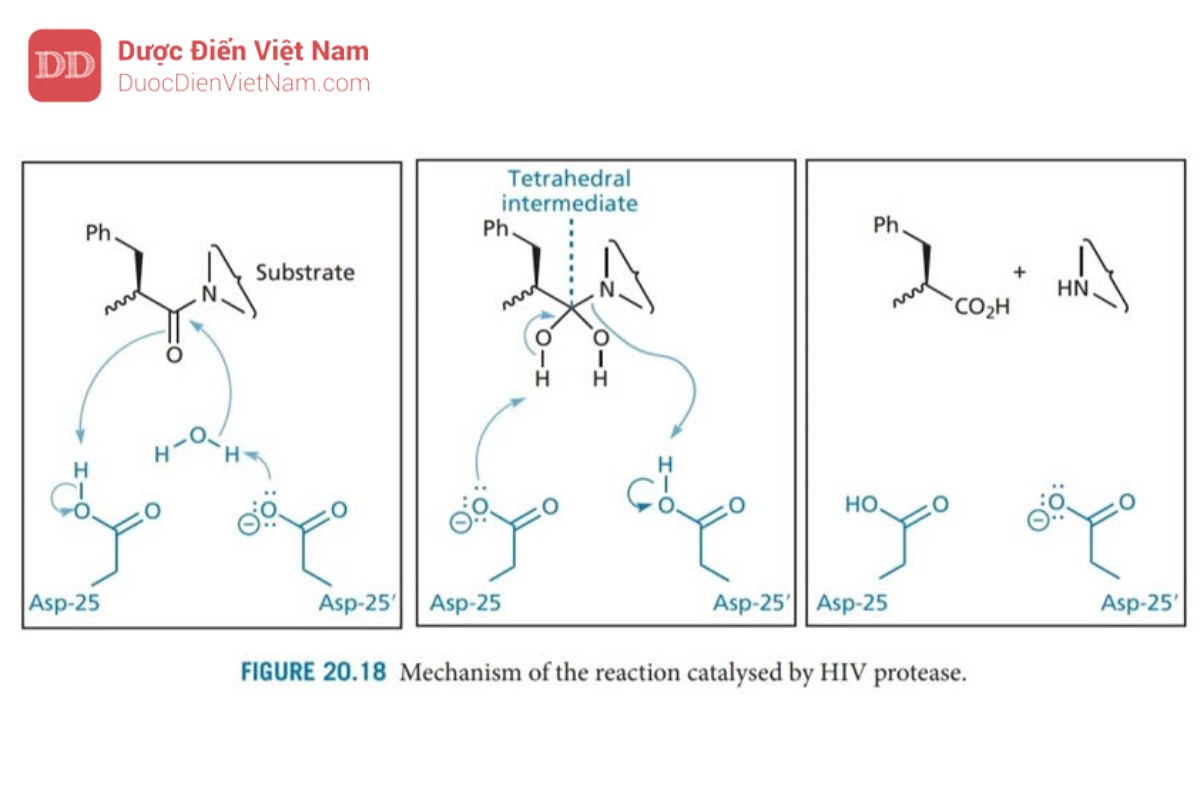
Các axit aspartic Asp-25 và Asp-25′ trên mặt phẳng của trung tâm hoạt động có liên quan đến cơ chế xúc tác. Mỗi acid amin này thuộc một trong các tiểu đơn vị protein và nhóm chức carboxylate tương tác với phân tử nước bắc cầu trong cơ chế thủy phân (Hình 20.18)
Thiết kế thuốc ức chế protease HIV (PIs)
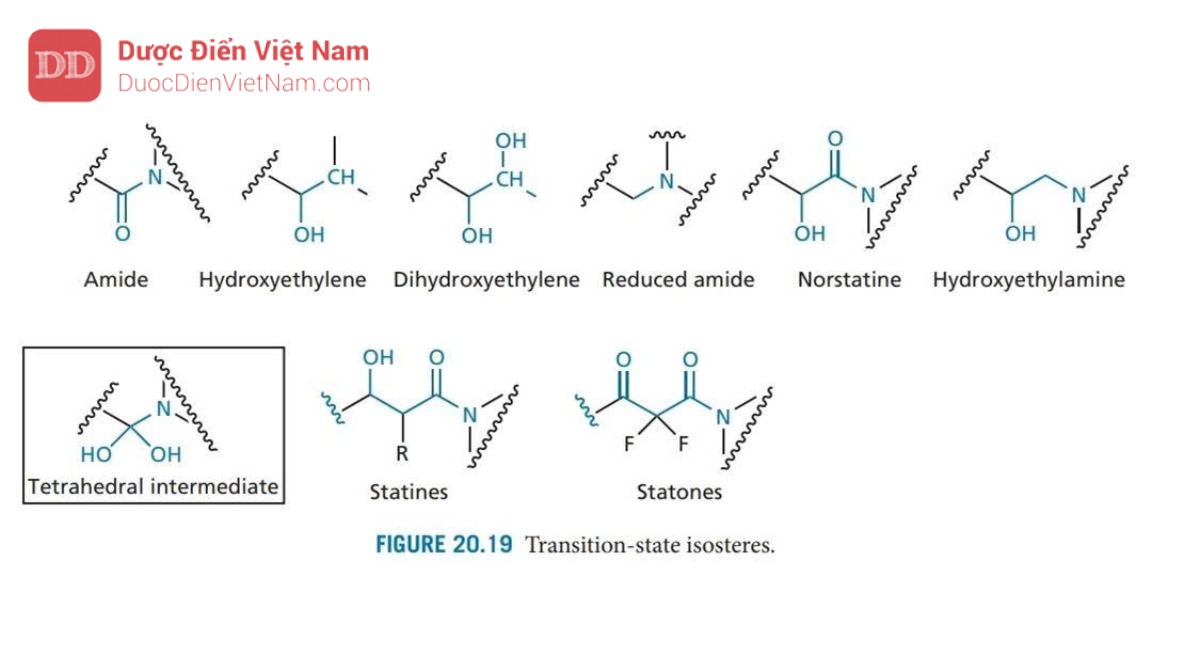
Một cơ chế thủy phân tương tự như trong Hình 20.18 diễn ra đối với aspartyl protease của động vật có vú được gọi là renin. Enzyme này đã được nghiên cứu rộng rãi trước khi phát hiện ra protease HIV và nhiều loại thuốc ức chế renin được thiết kế làm thuốc hạ huyết áp. Các chất này hoạt động như chất ức chế trạng thái chuyển tiếp và nhiều chiến lược phát triển thuốc mới từ chất ức chế renin đã được điều chỉnh phù hợp để thiết kế cấu trúc các chất ức chế enzyme Protease của HIV. Chất ức chế trạng thái chuyển tiếp được thiết kế để bắt chước trạng thái chuyển tiếp của phản ứng được xúc tác bởi enzyme. Ưu điểm của phương pháp này là trạng thái chuyển tiếp có khả năng tương tác với trung tâm hoạt động mạnh hơn so với cơ chất hoặc sản phẩm (cuối). Do đó, các chất ức chế có cấu trúc giống với trạng thái chuyển tiếp cũng có khả năng liên kết mạnh hơn. Trong trường hợp phản ứng được xúc tác bởi protease HIV, trạng thái chuyển tiếp có cấu trúc tứ diện như trong Hình 20.18. Do các cấu trúc như vậy không ổn định nên cần phải thiết kế chất ức chế có chứa nhóm đẳng cấu tại tâm tứ diện nhằm mục đích đảm bảo duy trì cấu trúc tứ diện nhưng đạt được sự ổn định trước quá trình thủy phân. May mắn thay, một số ứng viên đã được phát triển trong việc thiết kế các chất ức chế renin (Hình 20.19). Do đó, một số lượng lớn các cấu trúc mang nhóm đẳng cấu với trạng thái chuyển tiếp đã được tổng hợp, trong số các ứng viên này các chất mang nhóm đẳng cấu hydroxyethylamine tỏ ra có hiệu quả đặc biệt. Những ứng viên này mang nhóm hydroxyl bắt chước một trong các nhóm hydroxyl của trạng thái chuyển tiếp và liên kết với các gốc aspartate ở trung tâm hoạt động. Cấu trúc lập thể của nhóm này cũng quan trọng đối với hoạt tính, cấu hình R được ưu tiên hơn. Sự ưu tiên này được xác định bởi sự hiện diện của nhóm P1′.
Sau khi xác định được các nhóm đẳng cấu phù hợp, các chất ức chế được thiết kế dựa trên cơ chất peptide tự nhiên của enzyme vì chúng chứa các gốc axit amin tương thích với tám vị trí liên kết và đảm bảo liên kết tốt giữa cơ chất và enzyme. Về lý thuyết, có thể hợp lý khi thiết kế các chất ức chế sao cho tất cả tám vị trí liên kết đều được lấp đầy để thu được tương tác mạnh hơn. Tuy nhiên, điều này dẫn đến các cấu trúc có trọng lượng phân tử cao và do đó khả dụng sinh học qua đường uống kém. Do đó, hầu hết các PIs được thiết kế để đảm bảo liên kết tốt tại các vị trí cốt lõi trải dài từ S1 đến S1′. Sau đó, các nhóm thế khác được thêm vào ở hai đầu để phù hợp với các vị trí S2/S3 và S2′/S3′. Các chất ức chế sớm, chẳng hạn như saquinavir (Hình 20.21), có mạch nhánh axit amin liên kết với hầu hết các vị trí con từ S3 đến S3′. Thật không may, các hợp chất này có trọng lượng phân tử lớn và đặc tính peptide cao dẫn đến đặc tính dược động học kém. Các chất ức chế gần đây hơn đã được thiết kế với khả năng hòa tan trong nước và khả dụng sinh học qua đường uống tăng lên bằng cách sử dụng nhiều nhóm P2 và P2′ mới làm giảm trọng lượng phân tử và đặc tính peptide của các hợp chất. Các vị trí S2 và S2’ của enzyme protease dường như chứa cả axit amin phân cực (Asp-29, Asp-30) và kỵ nước (Val-32, Ile-50, Ile-84), cho phép thiết kế các loại thuốc chứa các nhóm P2 kỵ nước cũng có khả năng liên kết hydro. Người ta cũng có thể thiết kế một nhóm P1 có thể tương tác ở cả vị trí S1 và S3, cho phép loại bỏ một phần P3, do đó làm giảm trọng lượng phân tử. Nhóm P2 thường được gắn với P1 bằng liên kết acyl, vì oxy cacbonyl đóng vai trò là chất nhận liên kết hydro quan trọng với phân tử nước cầu nối được mô tả trước đây (Hình 20.17). Bây giờ chúng ta sẽ xem xét các chiến lược này được sử dụng như thế nào để thiết kế các PIs riêng lẻ
Saquinavir
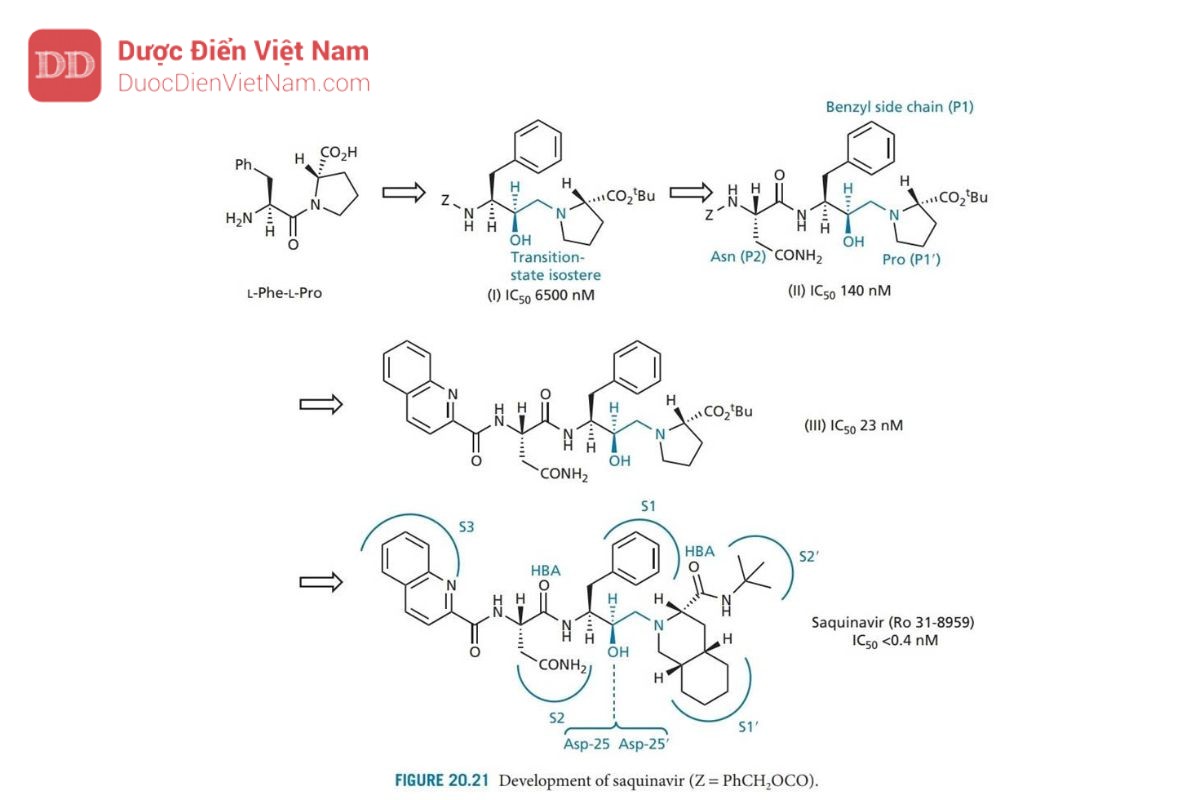
Saquinavir được Roche phát triển và là PIs đầu tiên xuất hiện trên thị trường, nó đóng vai trò là chuẩn mực cho tất cả các PIs khác. Thiết kế của saquinavir bắt đầu bằng cách xem xét cơ chất polypeptide của virus (pol, xem phần 20.7.2) và xác định vùng của polypeptide chứa liên kết peptide phenylalanine-proline. Trình tự pentapeptide Leu–Asn–Phe–Pro–Ile đã được xác định và dùng làm cơ sở cho việc thiết kế chất ức chế. Liên kết peptide thường bị thủy phân trong trình tự này là giữa Phe và Pro, và do đó liên kết này được thay thế bằng nhóm đẳng cấu: hydroxyethylamine để tạo ra cấu trúc ức chế thành công enzyme ( Hình 20.20 ). Các nhóm thế của axit amin của Leu–Asn–Phe– Pro–Ile được giữ lại trong cấu trúc này và liên kết với năm vị trí con S3–S2′. Mặc dù vậy, khả năng ức chế enzyme tương đối yếu. Hợp chất này cũng có trọng lượng phân tử cao và đặc tính giống peptide, cả hai đều làm giảm sinh khả dụng qua đường uống.
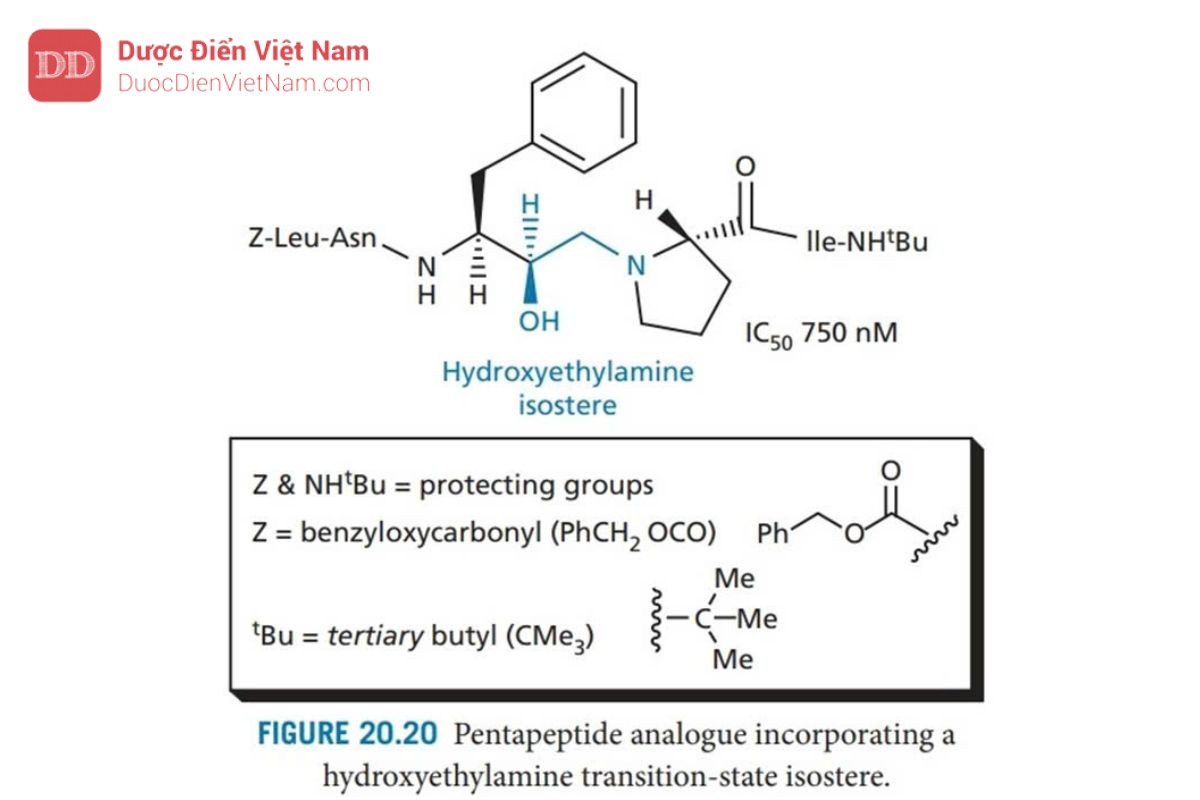
Do đó, nhóm nghiên cứu của Roche bắt đầu tìm kiếm một chất ức chế nhỏ hơn, bắt đầu từ cơ chất đơn giản nhất dipeptide Phe–Pro (Hình 20.21). Liên kết peptit được thay thế bằng nhóm đẳng cấu hydroxylamine và kết quả là liên kết N-C của cấu trúc (I) được bảo vệ bằng và phát hiện có hoạt tính ức chế yếu. Việc đưa nhóm asparagine (cấu trúc II) chiếm vị trí con S2 dẫn đến hoạt động tăng gấp 40 lần, điều đó có nghĩa là cấu trúc II mang hoạt tính mạnh hơn cấu trúc pentapeptide (Hình 20.20). Đây có vẻ là một kết quả ngoài dự kiến vì pentapeptide chiếm nhiều vị trí liên kết hơn. Tuy nhiên, người ta phát hiện ra rằng sự tương tác quan trọng của các chất ức chế nằm ở vùng lõi từ S2–S2′. Nếu việc bổ sung các nhóm được thiết kế để liên kết với các vị trí khác làm suy yếu sự tương tác với các vị trí cốt lõi, điều đó có thể dẫn đến sự suy giảm hoạt tính. Ví dụ, việc bổ sung leucine vào cấu trúc II dẫn đến giảm hoạt tính, mặc dù thực tế là leucine có thể chiếm vị trí con S3.
Cấu trúc II được sử dụng làm hợp chất dẫn đường mới và các gốc P1 và P2 được thay đổi để tìm ra nhóm tối ưu cho vị trí con S1 và S2. Kết quả, nhóm benzyl và mạch nhánh asparagine đã là những nhóm tối ưu. Một nghiên cứu về tinh thể bằng nhiễu xạ tia X của phức hợp chất ức chế_enzyme cho thấy nhóm bảo vệ (Z) chiếm vị trí S3, được chứng minh là một túi kỵ nước lớn. Do đó, nhóm bảo vệ được thay thế bằng hệ vòng quinoline lớn hơn nhằm khai thác tối ưu hơn phần không gian của túi thân dầu. Sự thay đổi này tạo ra hợp chất mới có hoạt tính tăng gấp sáu lần (cấu trúc III).
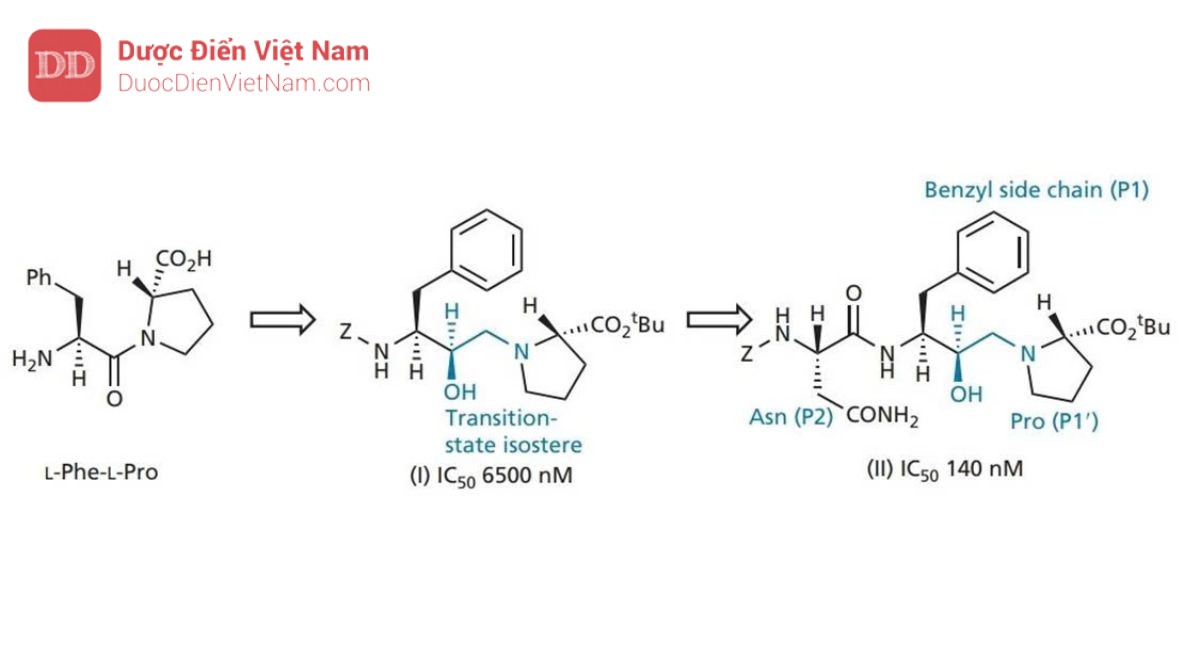
Những thay đổi cũng được thực hiện trên nhóm carboxyl của phân tử. Proline vừa khít với túi S1′, nhưng có thể thay thế nó bằng hệ vòng decahydroisoquino cồng kềnh hơn. Nhóm bảo vệ t-butyl ester được tìm thấy chiếm vị trí con S2′ và có thể được thay thế bằng nhóm t -butylamide được chứng minh là ổn định hơn trong các nghiên cứu trên động vật. Cấu trúc thu được (saquinavir) cho thấy hoạt tính tăng thêm 60 lần. Cấu hình R của nhóm hydroxyl ở trạng thái chuyển tiếp là rất cần thiết. Nếu cấu hình là S thì hợp chất sẽ mất hoạt tính.
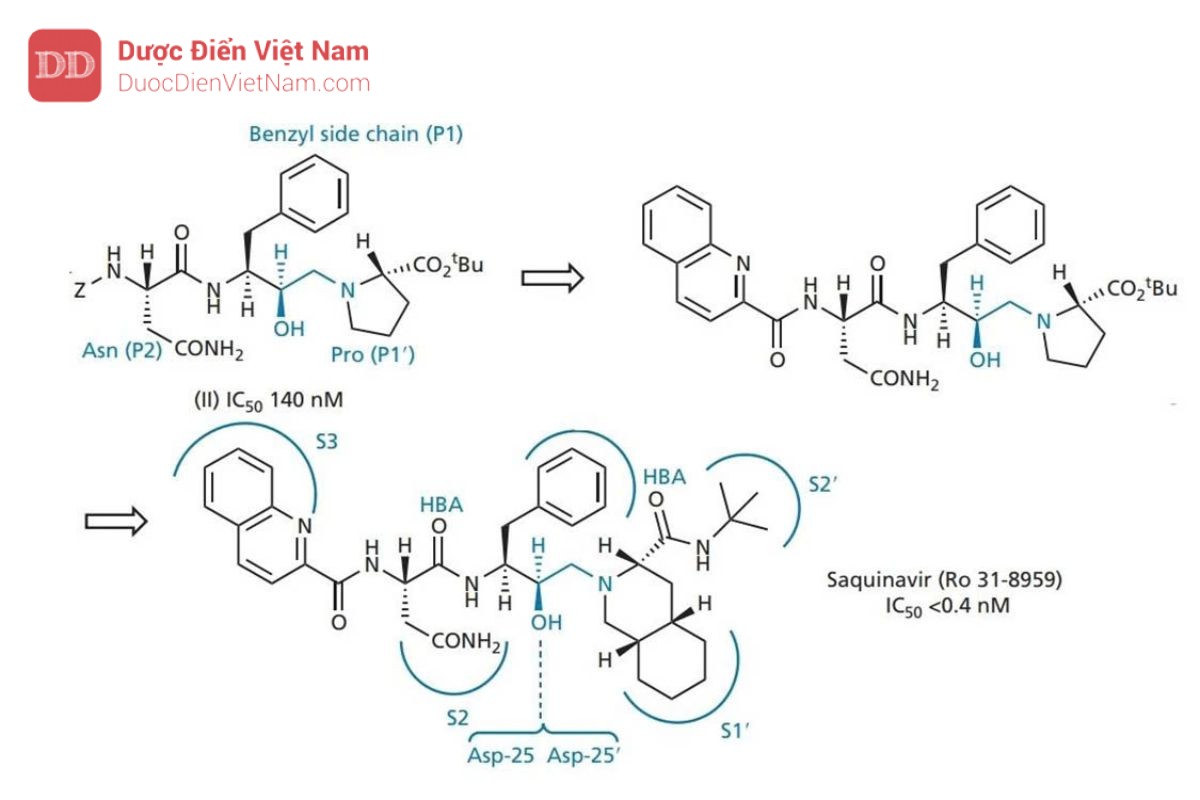
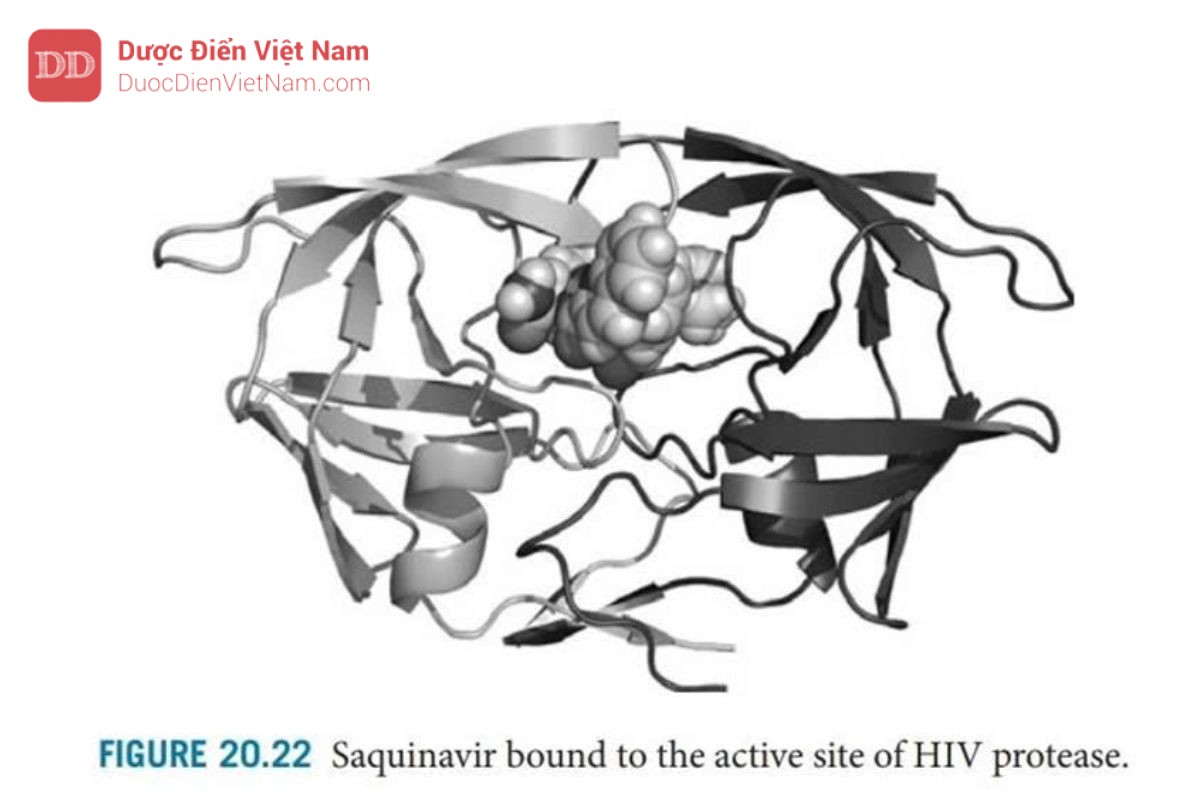
Cấu trúc tinh thể của phức hợp enzyme-saquinavir (Hình 20.21 và 20.22 ) đã chứng minh:
• các nhóm thế của thuốc chiếm 5 vị trí liên kết S3–S2′;
• nitơ t -butylamine được định vị theo cách mà các nhóm thế N tiếp theo sẽ không thể tiếp cận được vị trí con S3′;
• Tồn tại liên kết hydro giữa nhóm hydroxyl của nhóm hydroxyethylamine và aspartate tại trung tâm hoạt động của enzyme (Asp-25 và Asp-25′);
• Các nhóm carbonyl ở hai bên của chất ức chế đóng vai trò là chất nhận liên kết hydro với phân tử nước bắc cầu. Loại thứ hai hình thành liên kết hydro với các nhóm isoleucine trong vùng nắp của enzyme theo cách tương tự như trong hình 20.17.
Saquinivir vẫn được sử dụng trên lâm sàng nhưng có sinh khả dụng đường uống kém và dễ bị kháng thuốc. Nhiều nỗ lực khác nhau đã được thực hiện để thiết kế các chất tương tự đơn giản hơn của saquinavir, có trọng lượng phân tử thấp hơn, ít đặc tính peptide hơn để cải thiện sinh khả dụng đường uống.
Ritonavir và lopinavir
Ritonavir được Abbott Pharmaceuticals phát triển để tận dụng các đặc tính đối xứng của enzyme protease và trung tâm hoạt động của nó. Bởi vì trung tâm hoạt động có tính đối xứng C2, nên cơ chất có khả năng liên kết theo chiều ‘trái sang phải’ hoặc theo chiều ngược lại vì các vị trí liên kết S1–S4 giống hệt với các vị trí liên kết S1′–S4′. Điều này ngụ ý rằng có thể thiết kế các chất ức chế có tính đối xứng C2 có thể có một số ưu điểm.
+ Thứ nhất, các chất ức chế đối xứng sẽ thể hiện tính chọn lọc cao hơn đối với protease của virus so với protease aspartyl của động vật có vú (có trung tâm hoạt động bất đối xứng).
+ Thứ hai, các phân tử đối xứng có thể làm cho peptidase khó nhận biết hơn dẫn đến sinh khả dụng đường uống được cải thiện.
+ Thứ ba, sự phát triển của saquinavir cho thấy acid amin mang nhóm benzyl cho liên kết tối ưu tại S1. Vì vị trí con S1′ giống hệt với S1, nên với chất ức chế có cấu trúc đối xứng mang nhóm benzyl sẽ phù hợp với cả vị trí S1 và S1′ do vậy liên kết sẽ mạnh hơn dẫn đến hoạt tính được tăng cường. Tính Đối xứng cũng có thể được mở rộng cho các nhóm liên kết phù hợp tại các vị trí S2/S2′, v.v. Vì không có hợp chất dẫn đường nào có tính đối xứng C2 phù hợp với tính đối xứng của trung tâm hoạt động nên cần phải thiết kế một hợp chất dẫn đường. Điều này được thực hiện bằng cách xem xét sản phẩm trung gian mang cấu trúc tứ diện có nguồn gốc từ chuyển hóa cơ chất tự nhiên. Người ta giả định rằng trục đối xứng C2 của trung tâm hoạt động cũng đi qua trung tâm phản ứng của chất trung gian này (Hình 20.23 ).
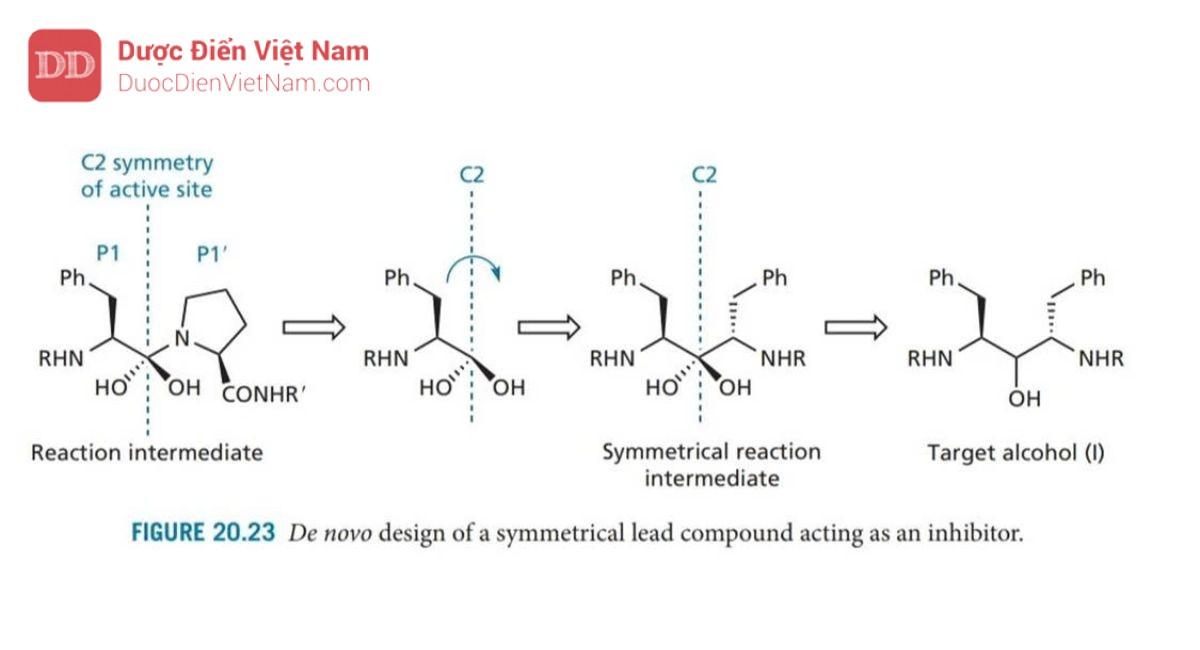
Vì nhóm benzyl được biết là tối ưu để liên kết với vị trí S1 nên phần bên trái của phân tử được giữ lại và phần bên phải bị loại bỏ. Sau đó, phần bên trái được quay sao cho hai gốc benzyl đối xứng với trục C2. Diol thu được vốn không ổn định, do đó một trong các nhóm OH ancol bị loại bỏ ta thu được ancol có cấu trúc (I) (I; R = H). Để kiểm tra xem phân tử mục tiêu này có phù hợp với tính đối xứng C2 của trung tâm hoạt động khi liên kết hay không, một thí nghiệm để đánh giá cấu trúc ancol vừa thu được. Kết quả rất khả quan và do đó ancol mục tiêu đã được tổng hợp. Mặc dù nó không có hoạt tính kháng virus nhưng nó lại thể hiện hoạt tính yếu như một chất ức chế enzyme, điều đó có nghĩa là nó có thể đóng vai trò là hợp chất dẫn đường để phát triển các hợp chất tối ưu hơn. Đây được coi là thành công của các kỹ thuật mới trong việc thiết kế cấu trúc hợp chất dẫn đường (phần 17.15).
Giai đoạn tiếp theo là mở rộng phân tử để tận dụng các vị trí S2 và S2’. Một loạt các cấu trúc đã được tổng hợp và thử nghiệm, cho thấy khả năng ức chế enzyme được cải thiện đáng kể khi valine được thêm vào, và cải thiện hơn nữa khi valine có nhóm bảo vệ N (A 74704; Hình 20.24).
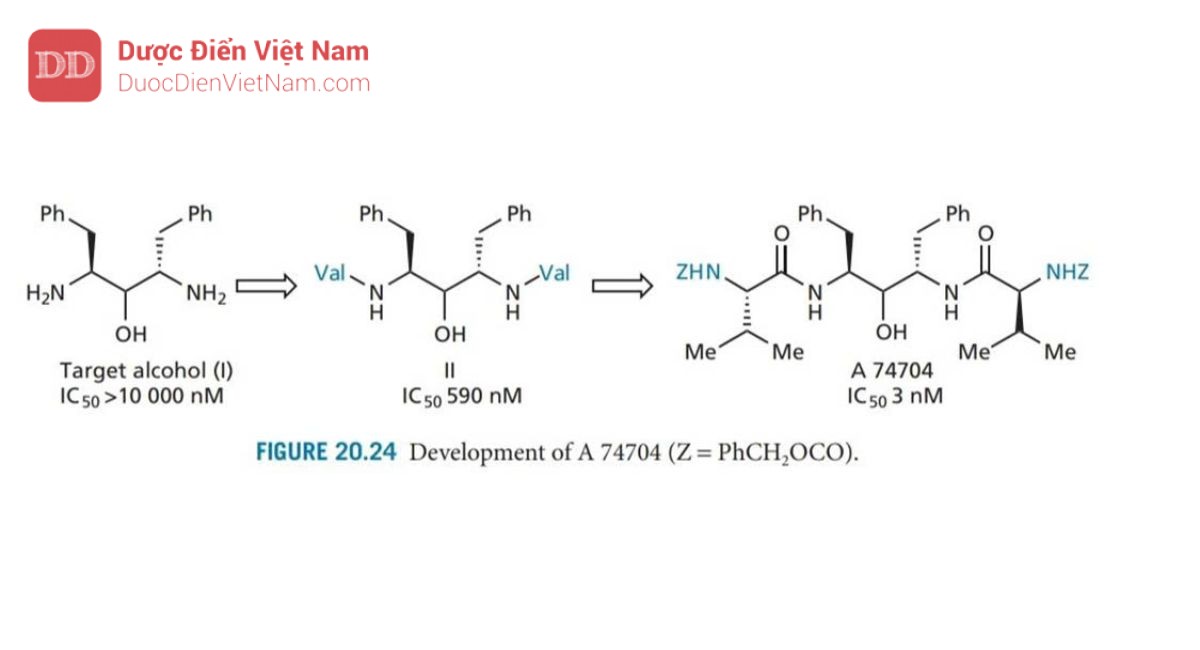
A 74704 cũng cho thấy hoạt tính kháng HIV invitro và có khả năng kháng lại sự phân giải protein.
Cấu trúc này được kết tinh đồng thời với enzyme protease tái tổ hợp và được nghiên cứu bằng phương pháp nhiễu xạ tia X để phát hiện ra mô hình đối xứng của liên kết hydro giữa chất ức chế và enzyme (Hình 20.25 ).
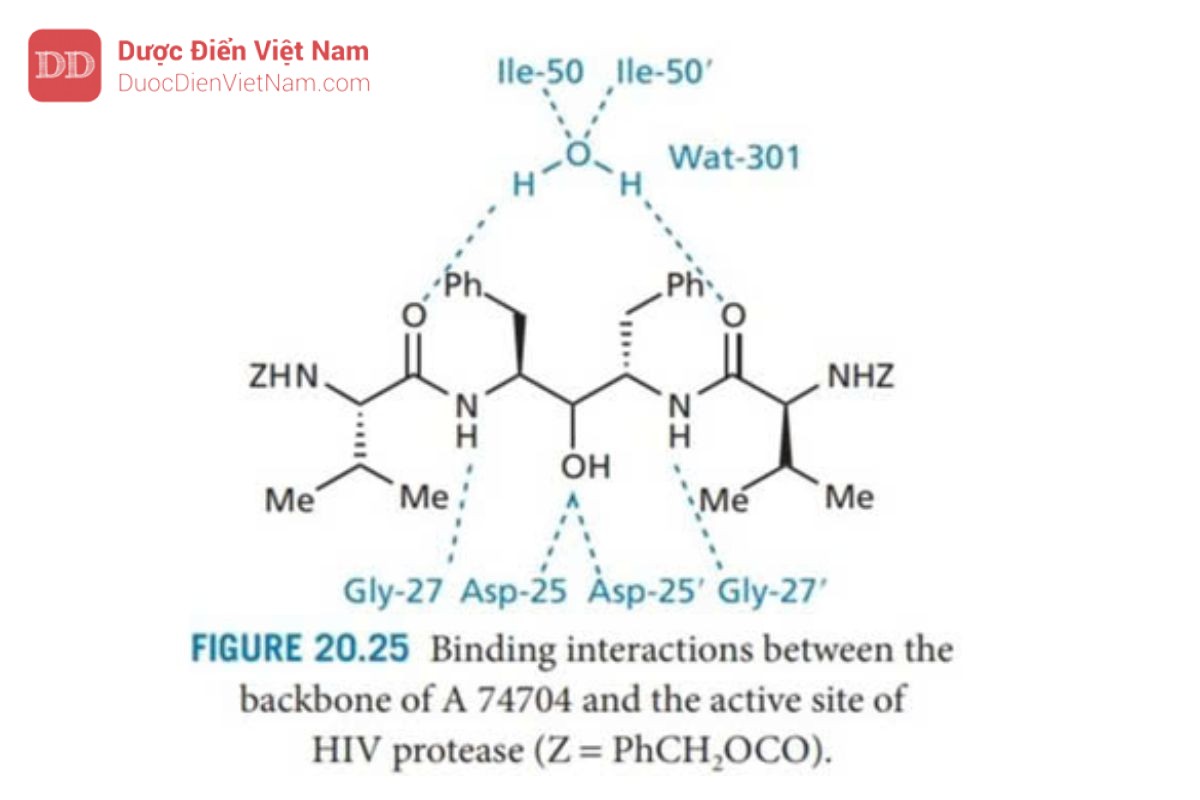
Người ta cũng phát hiện ra rằng một phân tử nước (Wat 301) đóng vai trò là cầu nối liên kết hydro giữa các nhóm carbonyl của P2 và P2′, và nhóm NH của Ile 50 và Ile-50′ trên nắp của enzyme. Trục đối xứng C2 của chất ức chế và trung tâm hoạt động đi qua nhau trong phạm vi 0,2 Å và lệch một góc chỉ 6°, thể hiện tính hợp lý của chiến lược thiết kế.
Phân tích sâu hơn về cấu trúc tinh thể cho thấy các nhóm NH trên chất ức chế liên kết với Gly-27 và Gly-27′, nhưng quá gần nhau để liên kết hydro phát huy hiệu quả. Để giải quyết vấn đề này, người ta quyết định thiết kế các chất ức chế đối xứng trong đó các nhóm NH có liên quan sẽ được phân tách bằng một liên kết bổ sung. Để đạt được điều này, trục đối xứng C2 được đặt xuyên qua tâm của liên kết peptid nhạy cảm. Theo đó, quá trình thiết kế được lặp lại để tạo ra diol như trong Hình 20.26 có thể là hợp chất dẫn đường. Cấu trúc Diol tương tự như ancol (I) được mô tả trước đây đã được tổng hợp và thử nghiệm. Thật kỳ lạ, người ta phát hiện ra rằng cấu hình tuyệt đối của các trung tâm diol ít ảnh hưởng đến hoạt tính và hoạt tính của các diol nhìn chung mạnh hơn so với các ancol tương ứng.
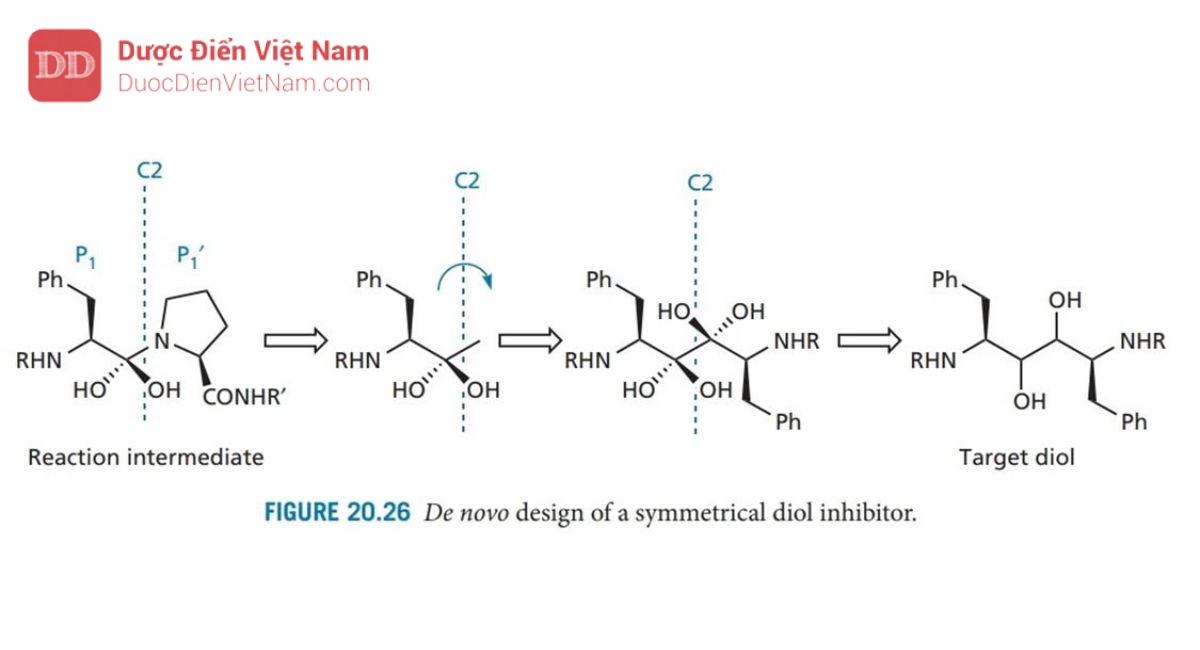
Ví dụ, diol tương đương với A 74704 (Hình 20.27) có hoạt tính mạnh gấp 10 lần. Thật không may, hợp chất này có độ tan trong nước kém, do đó cần phải tăng độ phân cực. Cấu trúc tinh thể của phức hợp chất ức chế enzyme cho thấy rằng các phần cuối cùng của phân tử được tiếp xúc với môi trường hòa tan, điều đó có nghĩa là có thể thêm nhiều nhóm phân cực hơn vào các vị trí đó mà không ảnh hưởng đến liên kết. Do đó, các nhóm phenyl cuối cùng được thay thế bằng các vòng pyridin có cực hơn. Các nhóm urethane gần các cực cũng được thay thế bằng các nhóm urê, dẫn đến A 77003 có khả năng hòa tan trong nước được cải thiện. Thật không may, sinh khả dụng đường uống vẫn chưa đạt yêu cầu và do đó cấu trúc này đã được đưa vào thử nghiệm lâm sàng dưới dạng thuốc kháng vi-rút tiêm tĩnh mạch.
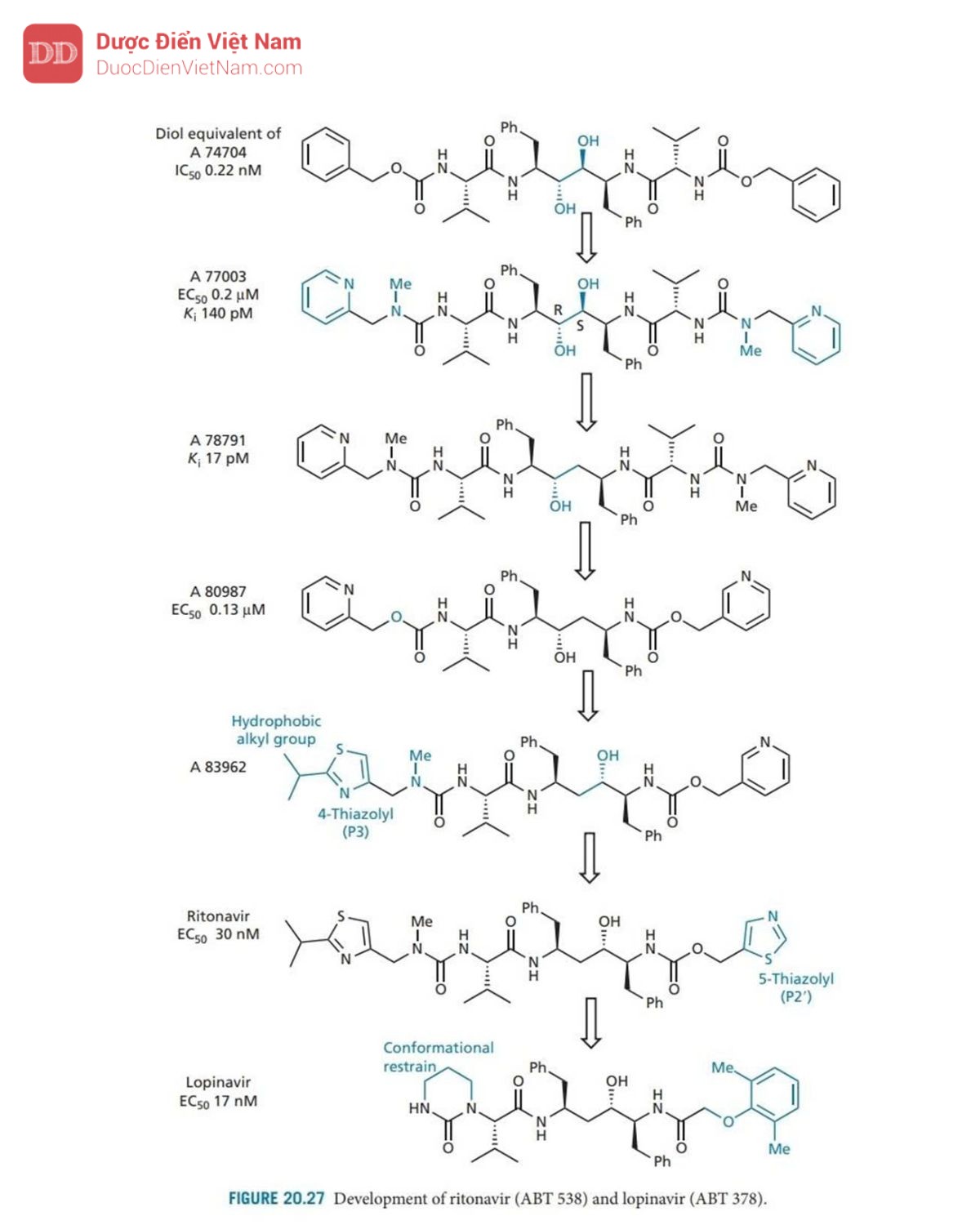
Các nghiên cứu mô hình về cách A 77003 có thể liên kết với trung tâm hoạt động đã gợi ý hai cách liên kết có thể có:
Cách 1: Mỗi nhóm hydroxyl diol hình thành liên kết hydro đối xứng với từng gốc aspartate
Cách 2: một trong số các nhóm hydroxyl liên kết hydro với cả hai nhóm aspartate.
Để nghiên cứu sâu hơn về vấn đề này, phương pháp nghiên cứu cấu trúc tinh thể nhiễu xạ tia X đã được thực hiện trên phức hợp chất ức chế enzyme, cho thấy rằng liên kết không đối xứng đang diễn ra, nhờ đó ( R )-OH tham gia liên kết hydro với cả hai acid amin aspartate và (S) -OH chỉ có thể tạo thành một liên kết hydro duy nhất. Phân tích cũng cho thấy rằng sự phân cực ngày càng tăng của NH amide không thể cải thiện các tương tác liên kết hydro với Gly-27 và Gly-27′. Vì vậy, sự cải thiện hoạt tính diol so với ancol (I) là do những lý do khác ngoài những lý do được đề xuất. Trên thực tế, hoạt động tốt hơn của các diol có thể là do sự liên kết tốt hơn của các nhóm P’ với các vị trí con S’.
Việc nhóm (S)-hydroxyl chỉ tạo ra một tương tác liên kết hydro gợi ý rằng nhóm S-hydroxyl nên loại bỏ vì năng lượng thu được từ chỉ một tương tác liên kết hydro nhỏ hơn năng lượng cần thiết để khử nhóm hydroxyl trước khi liên kết. Sau khi thực hiện loại bỏ nhóm thu được A 78791, có hoạt tính được cải thiện và được thể hiện qua tinh thể học tia X để liên kết theo cách tương tự như A 77003.
Sau đó, một nghiên cứu đã được thực hiện để đánh giá những thay đổi ảnh hưởng của kích thước phân tử, độ hòa tan trong nước và liên kết hydro đến dược động học và hoạt tính của các tác nhân này. Điều này tạo ra A 80987, trong đó valine P2’ bị loại bỏ và các nhóm urê ở gần cuối được thay thế bằng các nhóm urethane. Nhìn chung, người ta thấy rằng sự hiện diện của N -methylureas cải thiện độ tan trong nước và sinh khả dụng, trong khi sự hiện diện của urethanes (hoặc carbamate) giúp kéo dài thời gian bán hủy trong huyết tương và hiệu lực tổng thể. Do đó, chúng ta có thể điều chỉnh các đặc tính này bằng cách chọn nhóm phù hợp ở hai đầu của phân tử.
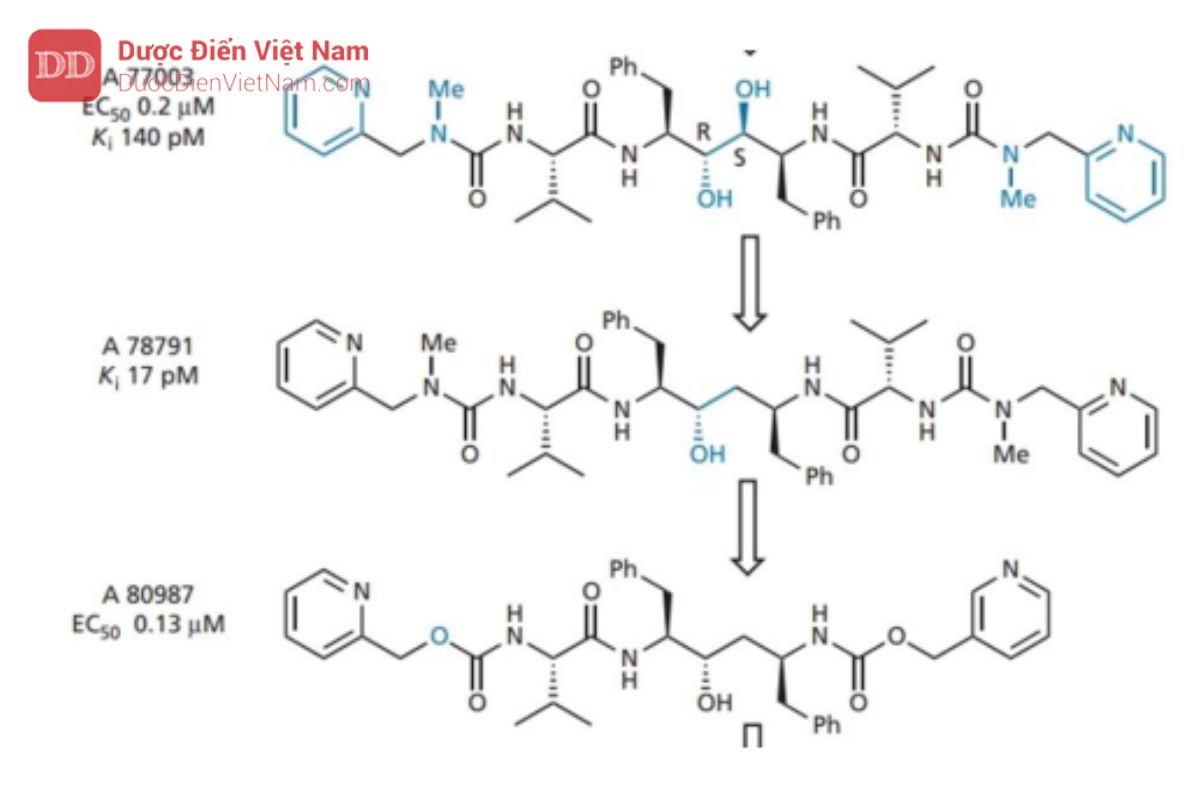
Mặc dù có ít tương tác hơn nhưng A 80987 vẫn duy trì hoạt tính và cải thiện sinh khả dụng qua đường uống. Tuy nhiên, nó có thời gian tồn tại trong huyết tương tương đối ngắn, liên kết mạnh với protein huyết tương và khó duy trì nồng độ điều trị cao. Các nghiên cứu về chuyển hóa sau đó cho thấy A80987 bị oxy hóa N ở một hoặc cả hai vòng pyridine và các chất chuyển hóa được bài tiết chủ yếu qua mật.
Trong nỗ lực chống lại điều này, nhiều chiến lược thiết kế khác nhau đã được thực hiện. Đầu tiên, các nhóm alkyl được đặt trên vòng pyridin ở vị trí ortho với nitơ. Những nhóm này được dự định hoạt động như một lá chắn không gian nhưng tỏ ra không hiệu quả trong việc ngăn chặn quá trình chuyển hóa. Sau đó người ta đề xuất giả thiết quá trình chuyển hóa có thể bị giảm đi nếu các vòng pyridin nghèo electron hơn và do đó các nhóm thế methoxy hoặc amino được thêm vào làm nhóm rút electron.
Tuy nhiên, điều này cũng không ngăn cản được quá trình chuyển hóa. Cuối cùng, vòng pyridine ở P3 được thay thế bằng nhiều loại dị vòng trong nỗ lực tìm ra một hệ thống vòng khác có thể hoạt động như một nhóm đẳng cấu sinh học nhưng ít nhạy cảm hơn với quá trình trao đổi chất. Kết quả tốt nhất thu được khi sử dụng vòng 4-thiazolyl nghèo electron hơn. Mặc dù khả năng hòa tan trong nước giảm nhưng nó có thể được phục hồi bằng cách đưa lại nhóm N -methylurea vào vị trí của một trong các
ure than. Những cải tiến hơn nữa về hoạt tính thu được bằng cách đặt các nhóm alkyl kỵ nước ở vị trí số 2 của vòng thiazole (P3) và sau đó thay đổi vị trí của nhóm hydroxyl trong trạng thái chuyển tiếp. Kết quả tạo ra A 83962, cho thấy hiệu lực tăng gấp tám lần so với A 80987.
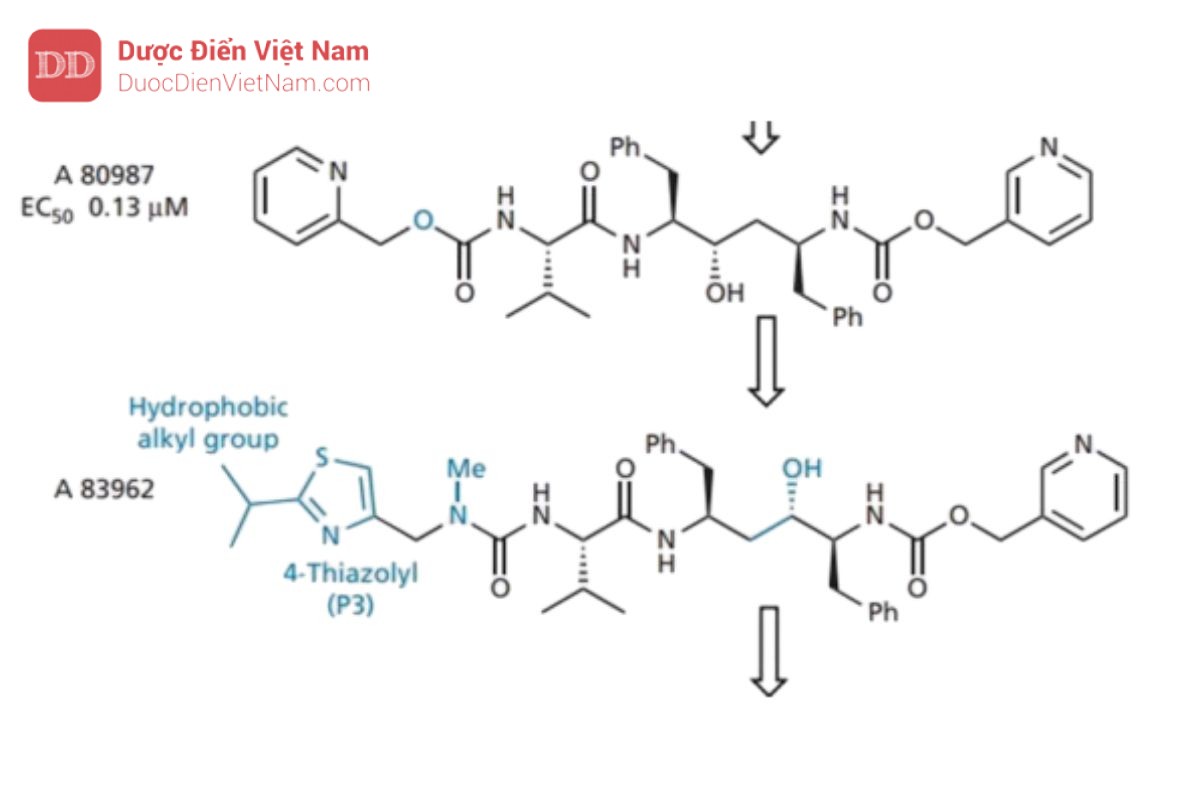
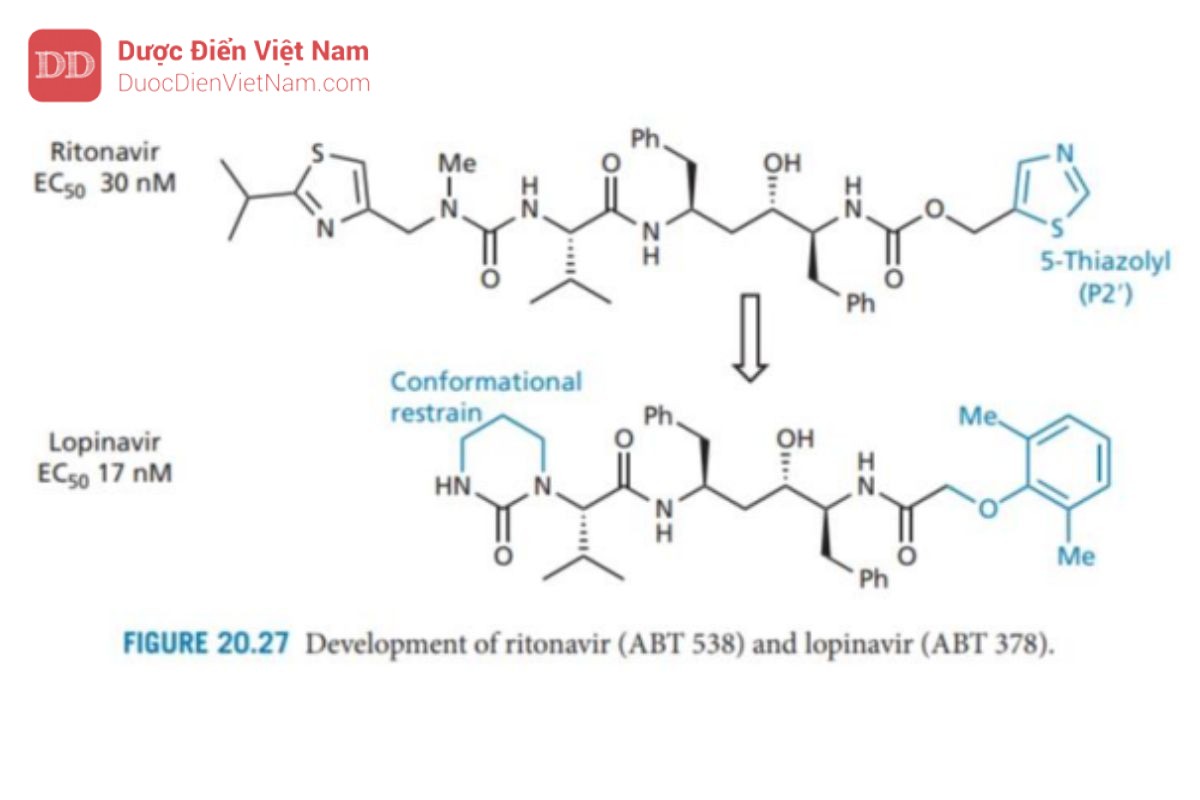
Bây giờ sự chú ý chuyển sang nhóm pyridine ở P2′ được thay thế bằng nhóm 5-thiazolyl để tạo ra ritonavir, có hoạt tính và sinh khả dụng qua đường uống tốt. Hoạt tính tốt có được do liên kết hydro hình thành giữa thiazolyl N và Asp-30 (cụ thể là nhóm NH+ ỏ cuối mạch peptide). Điều này phù hợp với tương tác liên kết hydro tương tự liên quan đến pyridine N trong A 80987. Sinh Khả dụng được<br />
cải thiện do ổn định hơn về chuyển hóa (ổn định hơn 20 lần so với A 80987) và có thể đạt được nồng độ điều trị trong huyết tương của thuốc kéo dài 24 giờ sau khi uống.
Các chủng vi rút kháng thuốc đã xuất hiện khi chỉ sử dụng ritonavir. Nguyên nhân do đột biến xảy ra thay thế valine (acid amin thứ 82 trên tiểu đơn vị protein của enzyme) thành alanine, threonine hoặc phenylalanine. Từ kết quả nghiên cứu tinh thể bằng phương pháp nhiễu xạ tia X cho biết tồn tại tương tác kỵ nước quan trọng giữa nhóm thế isopropyl trên nhóm thiazolyl P3 của ritonavir và nhóm thế isopropyl của Val-82 đã bị mất do đột biến này.
Sự phát triển thuốc tiếp theo đã dẫn đến lopinavir (Hình 20.27), trong đó nhóm thiazolyl P3 bị loại bỏ và nhóm urê đóng vòng được hợp nhất để cố định cấu hình. Điều này cho phép tăng cường tương tác liên kết hydro với vị trí S2, giúp cân bằng lại tương tác bị mất khi loại bỏ nhóm thiazolyl. Vì cấu trúc này không có bất kỳ tương tác nào với Val-82 nên nó có tác dụng chống lại chủng kháng ritonavir.
Indinavir
Merck đã thiết kế một PIs mạnh chứa nhóm đẳng cấu hydroxyethylene (L 685,434). Thật không may, nó sinh khả dụng thấp và độc với gan. Thời điểm này, các nhà nghiên cứu của Merck kết luận rằng có thể tận dụng được tính chất đối xứng của địa điểm đang hoạt động. Vì vị trí S và S′ đối xứng nhau nên có thể kết hợp một nửa PIs này với một nửa PIs khác để tạo ra chất ức chế khác biệt về mặt cấu trúc. Một nghiên cứu sàng lọc dựa trên mô hình đã được thực hiện để đánh giá giả thuyết và nhóm
nghiên cứu của Merck đã quyết định kết hợp nửa P′ của L 685434 với nửa P′ của saquinavir. Phần P’ của saquinavir được lựa chọn vì cải thiện độ tan và phần P’ của L 685434 lựa chọn vì không có đặc tính peptide. Cấu trúc lai thu được (L 704.486) có hoạt tính ức chế enzyme protease yếu nhưng sự có mặt của hệ vòng decahydroisoquinoline mang lại khả năng hòa tan trong nước và sinh khả dụng qua đường uống tốt hơn (15%), như dự kiến.
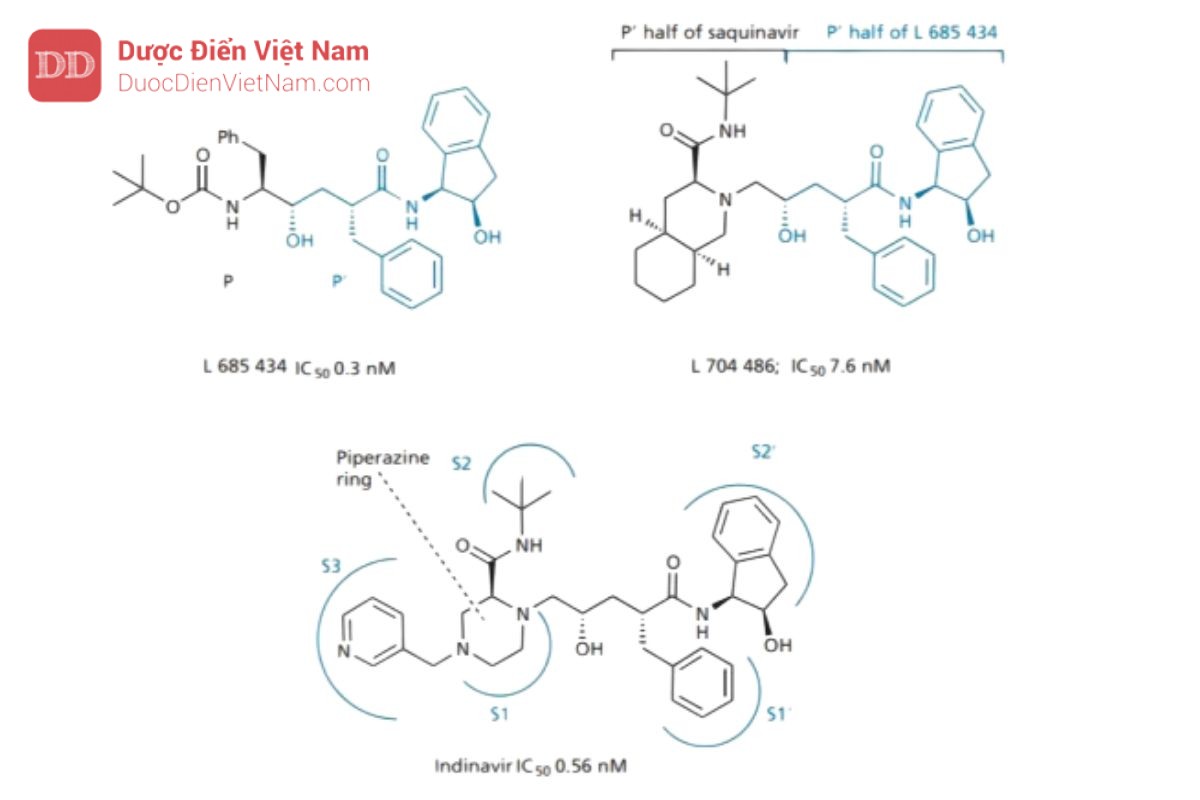
Những thay đổi tiếp theo nhằm mục đích cải thiện các liên kết với trung tâm hoạt động của enzyme, độ tan trong nước và sinh khả dụng qua đường uống. Vòng decahydroisoquinoline được thay thế bằng vòng piperazine, số lượng dị tố nitơ tăng giúp cải thiện khả năng độ tan trong nước và sinh khả dụng đường uống. Sau đó, nhóm thế pyridin được thêm vào khai thác vị trí S3 giúp cải thiện khả năng liên kết. Những thay đổi này tạo ra indinavir được đưa ra thị trường vào năm 1996.
Nelfinavir
Nelfinavir được phát triển bởi công ty Lilly, nhằm mục đích giảm trọng lượng phân tử và đặc tính peptide của PIs. AG1254 được sử dụng làm chất dẫn đường (Hình 20.29), hợp chất này chứa nhóm thế mở rộng ở P1 có khả năng liên kết với cả vị trí con S1 và S3 của enzyme. Loại nhóm P3 riêng biệt và cho phép thiết kế các hợp chất có trọng lượng phân tử thấp hơn. Họ cũng thiết kế một nhóm P2 mới thay thế acid amin asparagine đã có trong hợp chất dẫn đường. Nhóm này được thiết kế để liên kết hiệu quả với vị trí S2 và vì nó khác với bất kỳ acid amin nào trong tự nhiên nên đặc tính peptit của hợp chất giảm. Thật không may, hoạt tính kháng virus của AG 1254 không đủ mạnh và hợp chất này có độ tan trong nước kém.
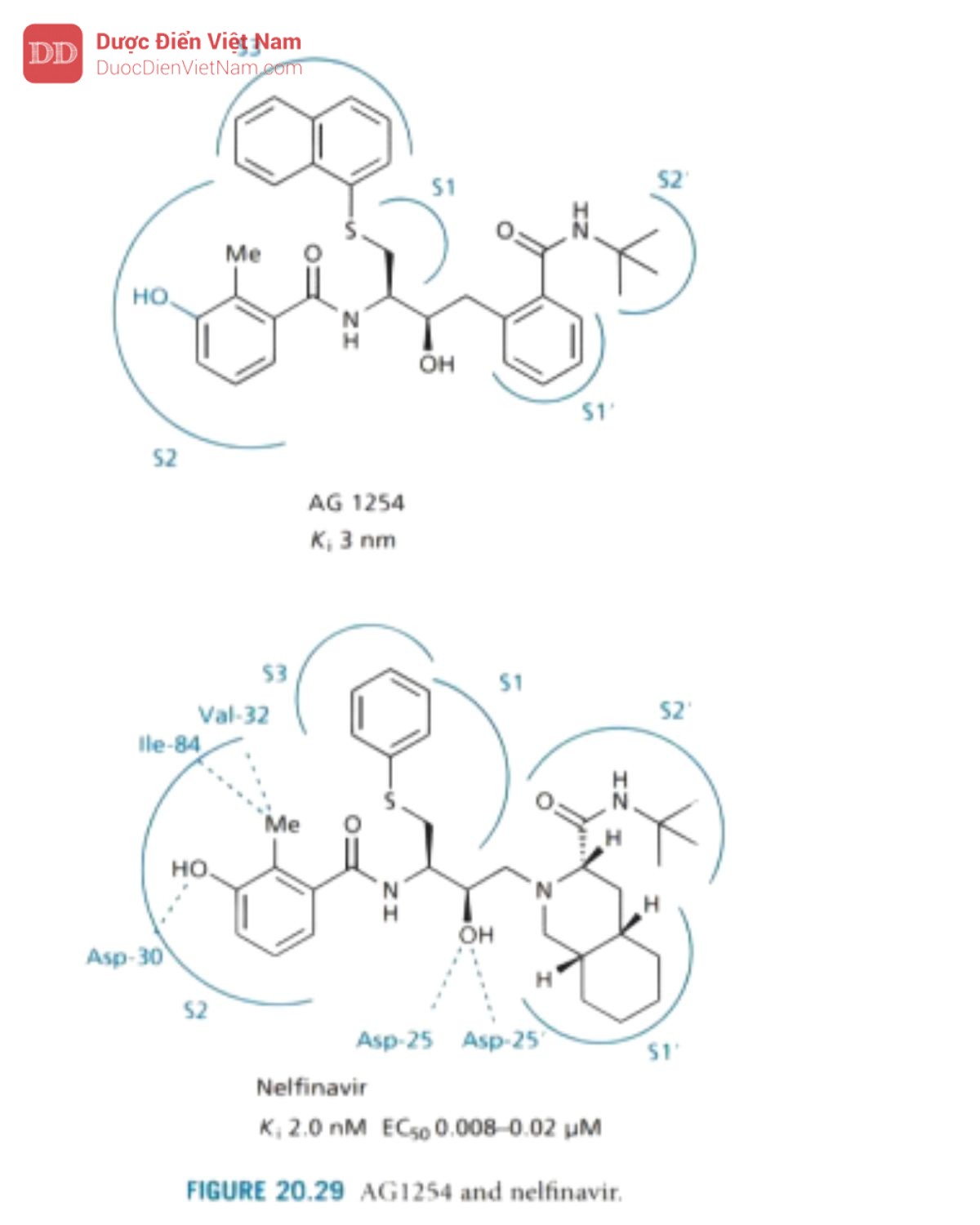
Công ty đã quyết định chuyển hướng và xem các nhóm thế được thiết kế mới của họ sẽ có tác dụng gì nếu chúng được kết hợp với saquinavir và kết quả cuối cùng tạo ra cấu trúc của nelfinavir. Nghiên cứu cấu trúc tinh thể của phức hợp nelfinavir enzyme cho thấy phân tử này được liên kết ở dạng cấu trúc mở rộng trong đó các tương tác liên quan đến khung phân tử tương tự như saquinavir. Một phân tử nước liên kết chặt chẽ đóng vai trò là cầu nối liên kết hydro giữa hai nhóm carbonylamid của chất ức chế và vùng nắp của enzyme, theo cách tương tự như các phức hợp chất ức chế enzyme khác. Cấu trúc tinh thể cũng cho biết nhóm S -phenyl định vị ở S1 và nhóm thế mở rộng tiếp cận vị trí S3. Hợp phần benzamide (chiếm túi S2) mang nhóm thế methyl tương tác với valine và isoleucine thông qua tương tác van der Waals và -OH phenol tương tác với Asp-30 bằng liên kết hydro.
Palinavir
Palinavir (Hình 20.30) là một chất ức chế đặc hiệu có hoạt tính ức chế mạnh các protease HIV-1 và HIV-2. Nửa phân tử phía bên trái hoặc nửa P của phân tử được thiết kế tương tự như saquinavir và phân tử này bắt chước trạng thái chuyển tiếp hydroxyethylamine tương tự. Phía bên phải (P′) khác biệt và được thiết kế bằng cách sử dụng chiến lược mở rộng nhóm thế giống với cách phát triển nelfinavir. Trong trường hợp này, nhóm thế P1’ được mở rộng để chiếm hai vị trí liên kết S1’ và S3’. Điều này đạt được bằng cách thay nhóm proline ban đầu ở P1′ bằng axit 4-hydroxypipecolinic và thêm nhóm thế chứa pyridine để xâm nhập vào vị trí S3′.
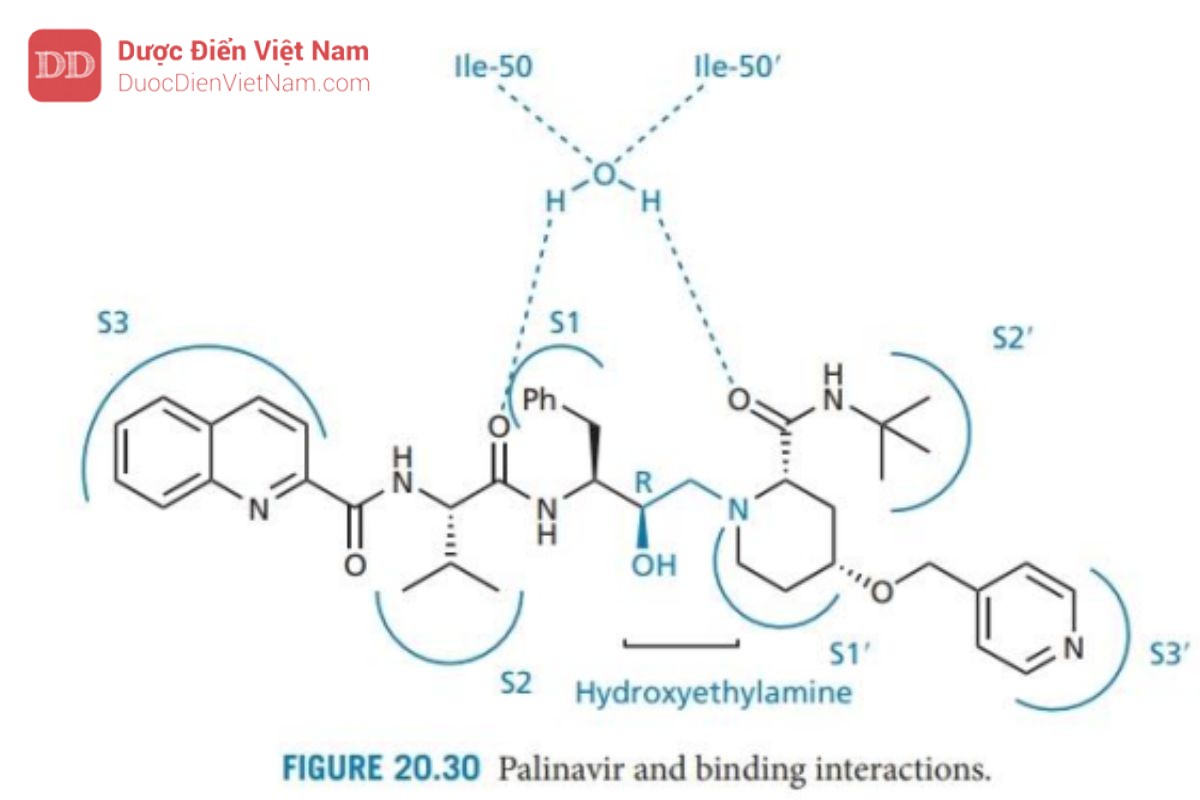
Nghiên cứu cấu trúc tinh thể của phức hợp enzyme-chất ức chế cho thấy các túi liên kết S3–S3′ đều tham gia tương tác. Hai nhóm carbonyl tương tác thông qua phân tử nước bắc cầu với các isoleucine trong các nắp enzyme. Nhóm hydroxyl tương tác với cả hai acid amin aspartate tại trung tâm hoạt động của enzyme protease. Cuối cùng, nguyên tử oxy và NH của tất cả các nhóm chức amid đều có khả năng liên kết hydro với các nhóm thế ở trung tâm hoạt động. Hướng phát triển tiếp theo là đơn giản hóa cấu trúc palinavir bằng cách gắn một nhóm duy nhất mở rộng trên hai vị trí liên kết, từ đó cho phép loại bỏ nhóm liên kết P3
Amprenavir và darunavir
Amprenavir (Hình 20.31) được thiết kế bởi Vertex Pharmaceuticals dưới dạng chất ức chế enzyme protease không có cấu trúc peptide sử dụng saquinavir làm hợp chất dẫn dường. Saquinavir có trọng lượng phân tử cao và đặc tính peptide cao, nên sinh khả dụng đường uống bị hạn chế. Do đó, hướng phát triển là thiết kế một chất tương tự có cấu trúc đơn giản hơn để giảm trọng lượng phân tử và mang ít đặc tính peptide hơn nhưng vẫn giữ được hoạt tính ức chế enzyme protease.
Đầu tiên, nhóm decahydroisoquinoline trong saquinavir được thay thế bằng nhóm isobutyl sulfonamide để tạo ra cấu trúc (I). Điều này có ưu điểm là giảm số lượng trung tâm bất đối xứng từ sáu xuống còn ba, cho phép tổng hợp dễ dàng hơn. Để tinh giản hơn nữa và giảm đặc tính peptid được thực hiện bằng cách thay nhóm P2 và P3 bằng carbamate tetrahydrofuran (THF), đây là nhóm mà trước đây nhóm nghiên cứu của Merck đã phát hiện là nhóm liên kết tốt cho vị trí S2. Cuối cùng, một nhóm amino được đưa vào nhóm phenylsulphonamide để tăng khả năng hòa tan trong nước và cải thiện mức độ hấp thu qua đường uống. Fosamprenavir là tiền chất phosphat của amprenavir.
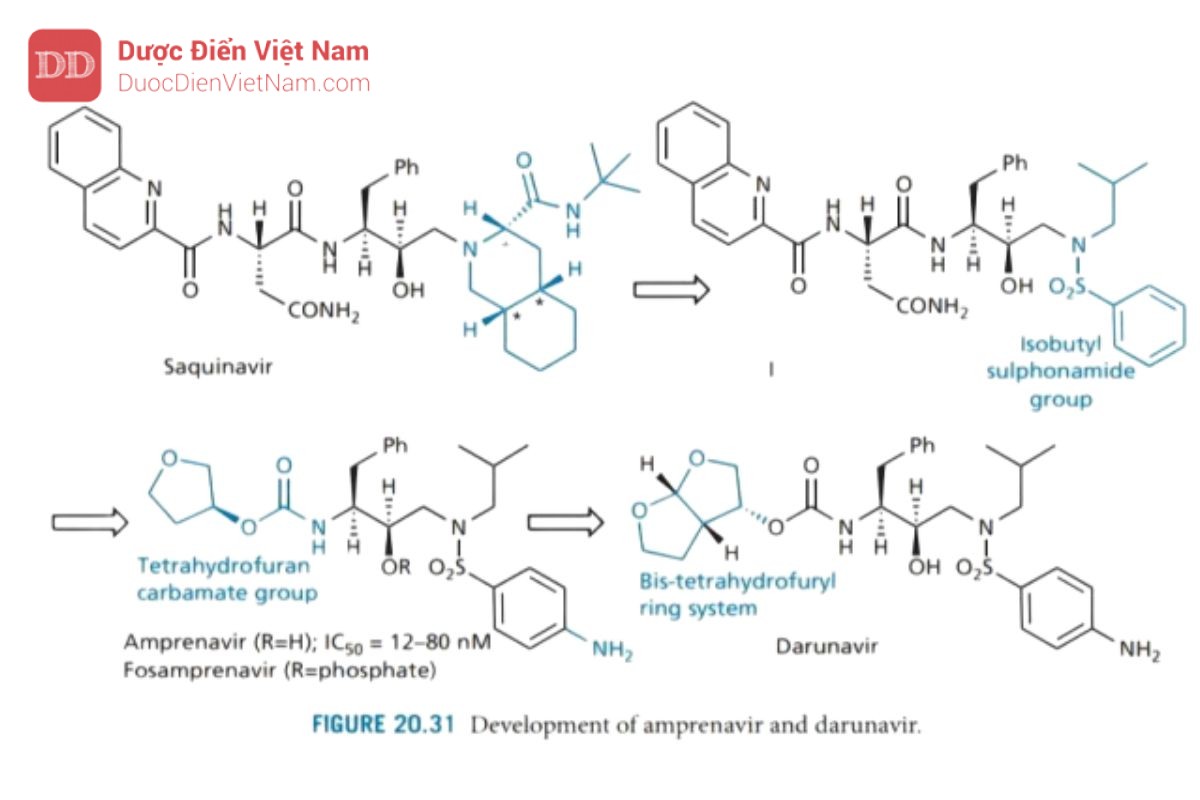
Nghiên cứu tiếp theo đã chỉ ra rằng hệ vòng bis-tetrahydro furyl ngưng tụ là nhóm liên kết với túi S2 kỵ nước tốt hơn đối so với THF đơn vòng vì nó khai thác tối ưu thể tích vùng không gian của túi S2 và hình thành các liên kết hydro giữa dị tố oxy trên vòng và các nhóm chức trên mạch peptide của enzym. Vì những tương tác này xảy ra với khung protein chứ không phải mạch nhánh của các axit amin nên đột biến ít có khả năng dẫn đến kháng thuốc. Darunavir là PIs thế hệ thứ hai có tính năng này, nhưng hiện tại cũng có một số hợp chất khác đang được nghiên cứu.
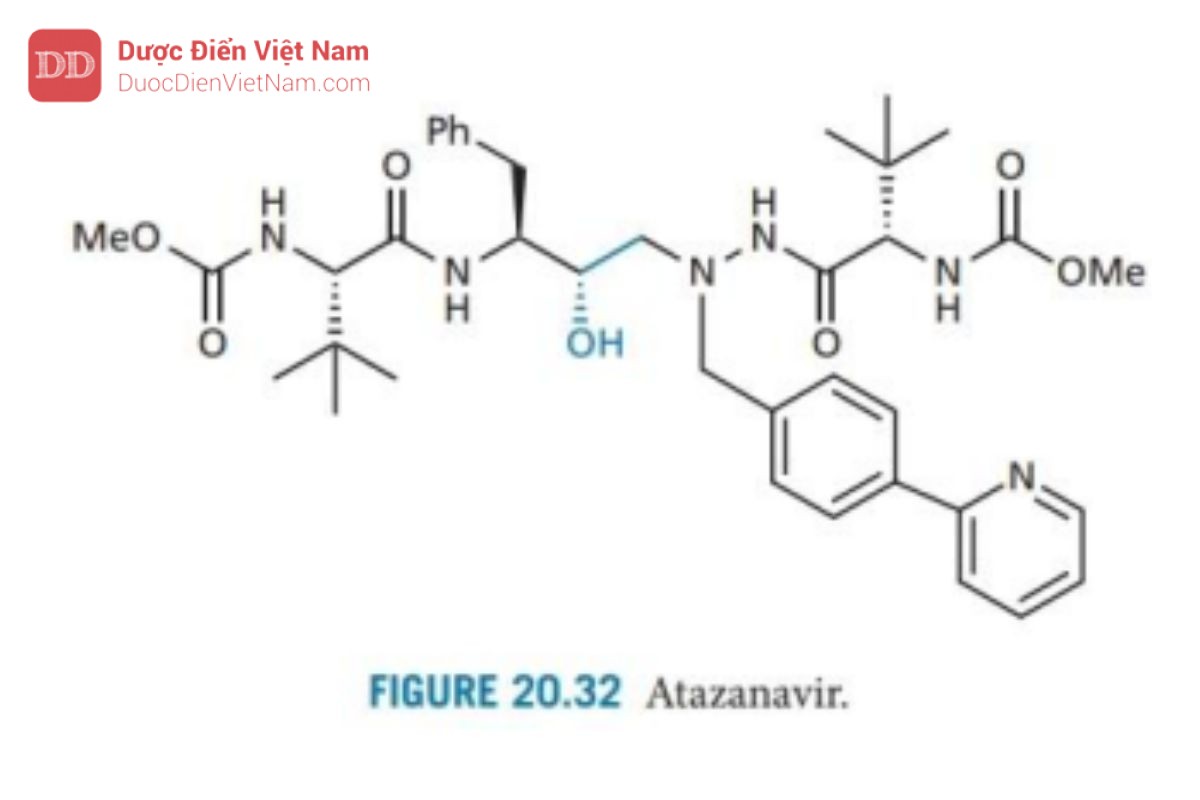
Atazanavir
Atazanavir (Hình 20.32) đã được phê duyệt vào tháng 6 năm 2003 là thuốc HIV-1 PIs tiên được sử dụng trong phác đồ phối hợp với chế độ liều dùng một lần mỗi ngày. Nó có cấu trúc tương tự như các hợp chất dẫn đường khi phát triển ritonavir. Nghiên cứu hiện tại đang xem xét khả năng sử dụng chất tương tự atazanavir có gắn đồng vị deuterium, được cho là có tốc độ chuyển hóa và bài tiết chậm hơn, đồng thời thời gian bán hủy tăng lên (xem thêm phần 14.2.4).
Tipranavir
Tipranavir (Hình 20.33) là một ví dụ về PIs được thiết kế từ hợp chất dẫn đường không có cấu trúc peptide. Việc sàng lọc số lượng lớn 5000 hợp chất có cấu trúc đa dạng đã dẫn đến phát hiện thuốc chống đông máu warfarin là một PIs yếu có hoạt tính kháng virus. Sau đó, nhiều chất tương tự warfarin khác nhau đã được thiết kế và thử nghiệm dẫn đến phát hiện ra rằng phenprocoumon (Hình 20.33) là một chất ức chế enzyme cạnh tranh mạnh hơn với hoạt tính kháng virus yếu. Cả hai cấu trúc này đều được sử dụng trong điều trị cho các mục đích khác và có sinh khả dụng đường uống cao. Do đó, chúng đóng vai trò là hợp chất dẫn đường đầy triển vọng cho các chất kháng virus không có cấu trúc peptit và mạng lại sinh khả dụng tốt qua đường uống.
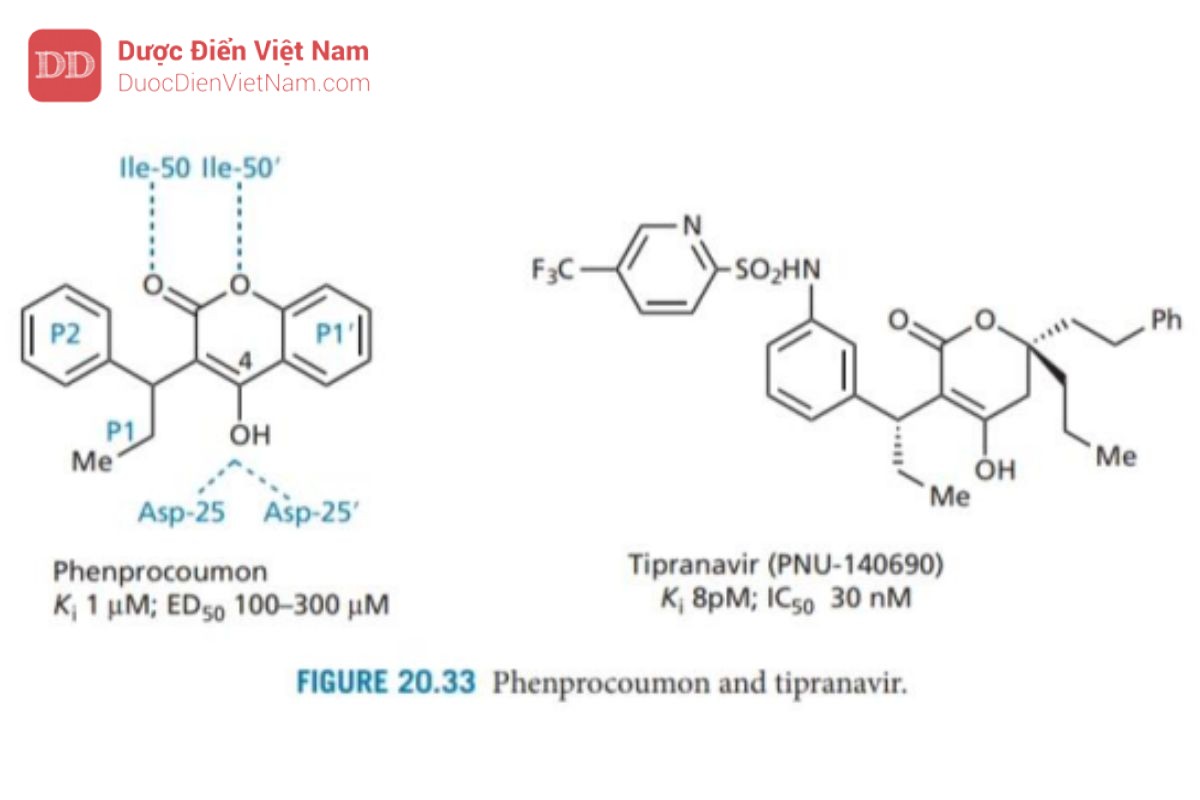
Cấu trúc tinh thể của phức hợp chất ức chế enzyme đã được xác định cho thấy nhóm 4-OH có thể hình thành liên kết hydro với gốc aspartate của enzymer, trong khi hai nguyên tử oxy ơt nhóm lacton có thể hình thành liên kết hydro trực tiếp với các nhóm isoleucine (Ile-50 và Ile -50′) trong các nắp enzyme. Không giống như tất cả các PIs trước đó, không có phân tử nước bắc cầu. Do đó, các hợp chất này đại diện cho một nhóm chất ức chế mới với dược điển mới về tương tác liên kết hydro. Cấu trúc tinh thể cũng cho thấy các nhóm ethyl và phenyl lần lượt phù hợp với các vị trí S1 và S2, trong khi vòng benzen của hệ vòng coumarin phù hợp với vị trí S1. Phenprocoumon được sử dụng làm hợp chất dẫn đường để phát triển hơn nữa và dẫn đến việc phát hiện ra tipranavir.
Chiến lược thiết kế thay thế cho thuốc kháng virus nhắm vào enzyme protease HIV
Một cách tiếp cận khác để ức chế enzyme protease là ngăn chặn sự hình thành của nó ngay từ đầu. Các nghiên cứu đang được tiến hành để thiết kế các chất ức chế liên kết protein-protein nhằm ngăn chặn sự liên kết của hai tiểu đơn vị protein tạo nên nó.
Một cách tiếp cận thú vị khác là thiết kế các tiền chất chứa các hợp chất gây độc chỉ được hoạt hóa bởi protease HIV. Các tiền chất này sẽ chứa một nửa cấu trúc như cơ chất của HIV-protease, sao cho độc tố này chỉ được giải phóng trong các tế bào bị nhiễm HIV. Độc tố sau đó sẽ tấn công các mục tiêu tế bào và loại bỏ các tế bào đó.
Chất ức chế các mục tiêu phân tử khác
Các chất kháng virus đang được phát triển theo hướng ngăn chặn việc sản xuất protein HIV Tat, protein cần thiết cho quá trình phiên mã của các gen khác của virus HIV. Trecovirsen (Hình 20.34) là một oligonucleotide photphorothioate chứa 25 nucleotide và được thiết kế để lai với mRNA có nguồn gốc từ gen HIV ngăn chặn sự dịch mã của nó thành protein HIV. Nó bị loại bỏ từ các thửnghiệm lâm sàng do độc tính nhưng một oligonucleotide tương tự (GEM92) với độ ổn định cao hơn hiện đang được thử nghiệm lâm sàng
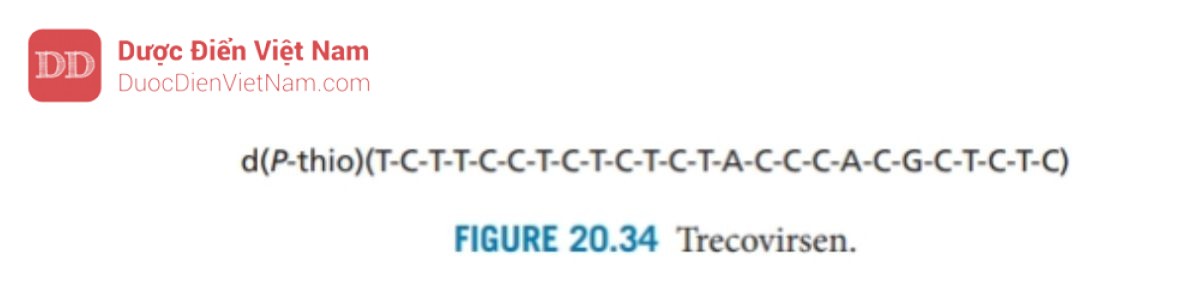
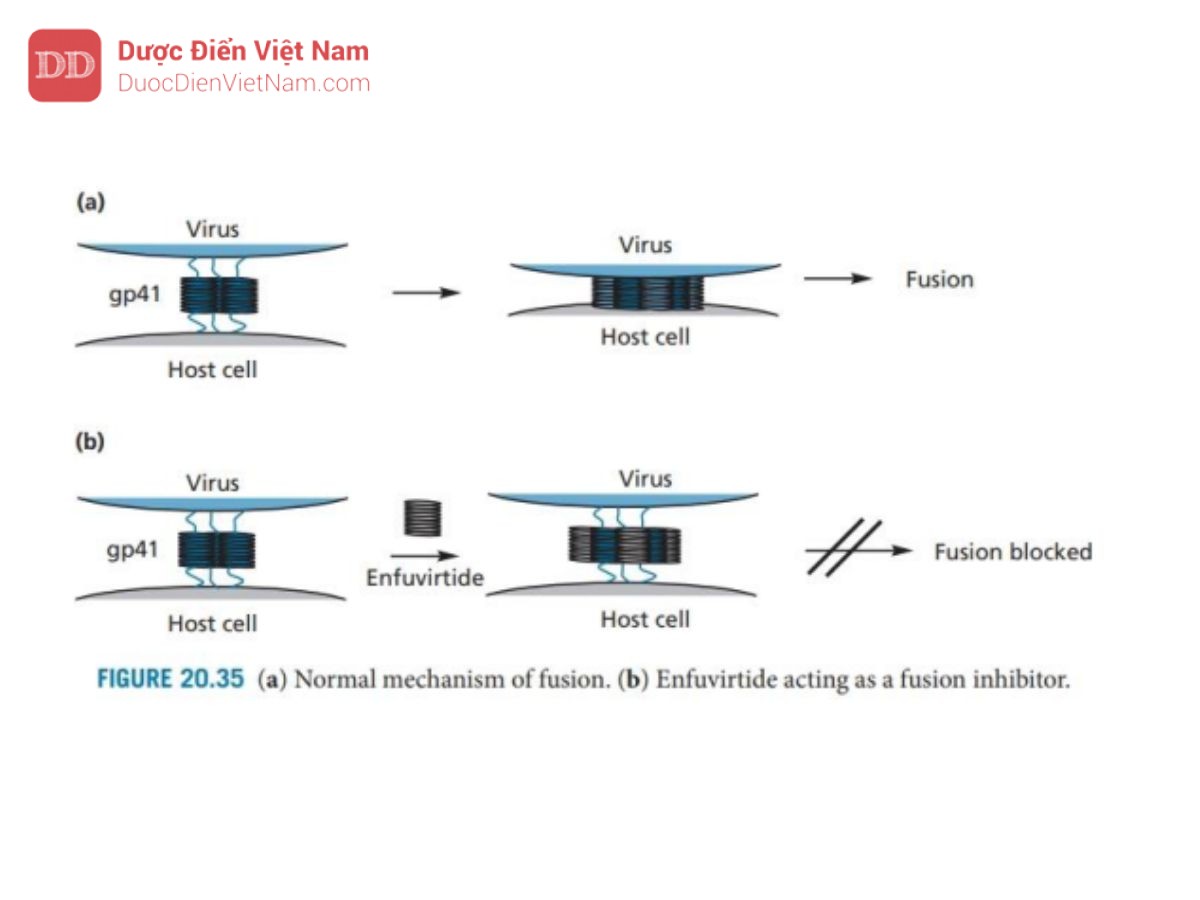
Các tác nhân khác đang được nghiên cứu để điều trị HIV bao gồm các chất ức chế integrase và các chất ức chế xâm nhập tế bào. Việc ngăn chặn sự xâm nhập của virus vào tế bào chủ là điều đặc biệt mong muốn vì nó còn rất sớm trong vòng đời. Enfuvirtide đã được phê duyệt vào tháng 3 năm 2003
với tư cách là thành viên đầu tiên của nhóm thuốc ức chế sự dung hợp. Nó là một polypeptide bao gồm 36 axit amin phù hợp với đầu C của protein virus gp41. Nó hoạt động bằng cách hình thành một chuỗi xoắn và liên kết với một nhóm gồm ba chuỗi xoắn tương tự thuộc protein gp41. Sự liên kết này ngăn chặn quá trình virus xâm nhập vào tế bào chủ. Để tạo ra sự dung hợp, protein gp41 sẽ neo virus vào màng tế bào của tế bào chủ. Sau đó, nó thay đổi về cấu hình trong đó virus sử dụng một nhóm gồm sáu chuỗi xoắn helix bằng cách sử dụng ba chuỗi xoắn đã ghim trên tế bào chủ làm tiêu điểm cho nhóm đó (Hình 20.35) rồi kéo màng của virion và tế bào chủ lại gần nhau từ đó tiến hành hợp nhất. Bằng cách liên kết với nhóm ba chuỗi xoắn trung tâm, enfuvirtide ngăn chặn sự hình thành hexamer cần thiết và ngăn chặn sự dung hợp.
Quá trình sản xuất enfuvirtide bao gồm 106 bước, khiến nó đắt tiền và có thể hạn chế sử dụng. Một hợp chất nhỏ hơn (BMS 378806) đang được nghiên cứu có khả năng liên kết với gp120 và ngăn chặn sự liên kết ban đầu của virus với CD4 trên bề mặt tế bào.
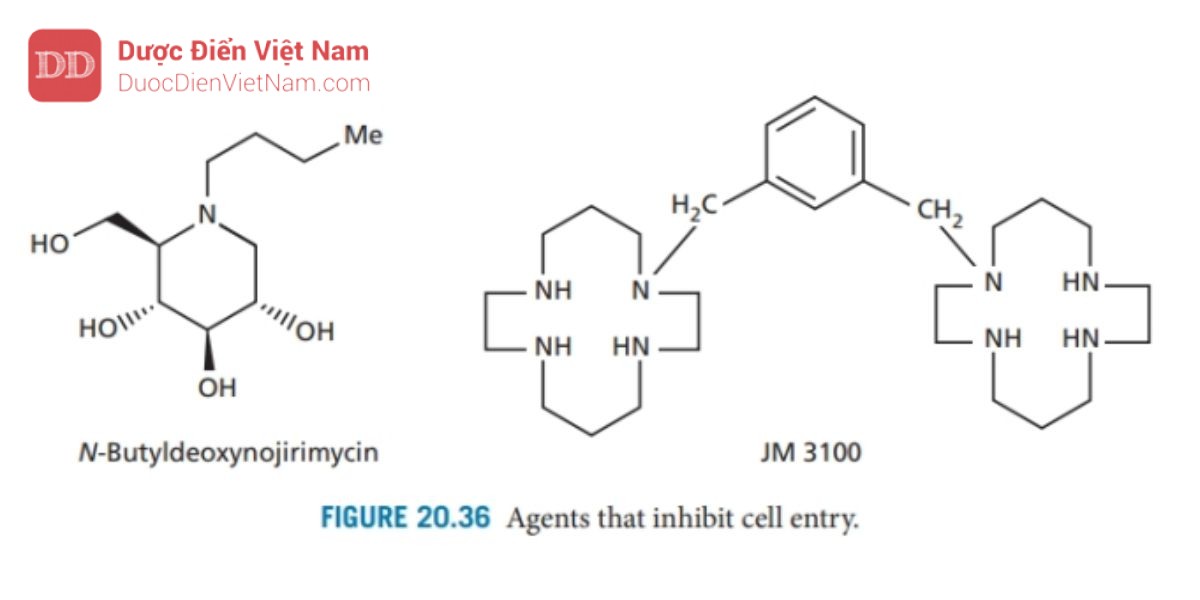
N-Butyldeoxynojirimycin (Hình 20.36) là một carbohydrat có tác dụng ức chế glycosidase – các enzyme xúc tác phân giải các phân tử carbohydrate liên kết với protein của virus. Nếu quá trình này bị ức chế, quá nhiều nhóm carbohydrate sẽ được gắn vào một protein, dẫn đến hình dạng protein bị thay đổi. Người ta cho rằng protein gp120 bị ảnh hưởng theo cách này và không thể tách ra như mô tả trong để lộ ra protein gp41 từ đó ngăn cản quá trình xâm nhập.
Bicyclams như JM 3100 (Hình 20.36) chặn thụ thể chemokine CCR5 và đang được nghiên cứu như một loại thuốc ngăn chặn sự dung hợp màng và sự xâm nhập của tế bào.
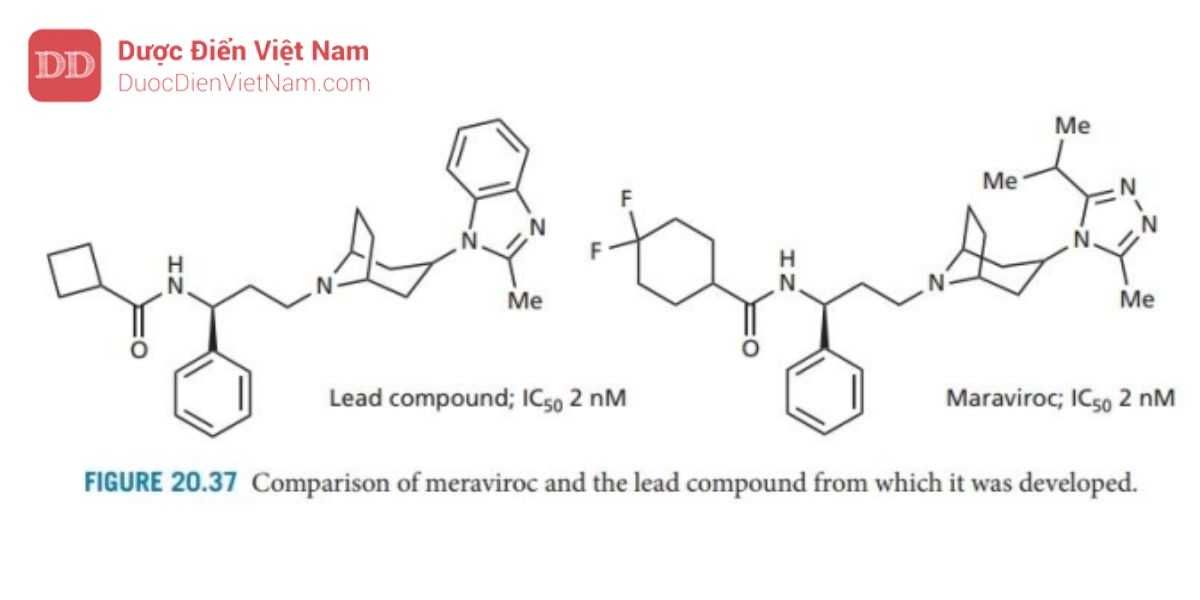
Maraviroc (Hình 20.37) đã được phê duyệt là chất đối kháng CCR5 vào năm 2007 và là tác nhân kháng HIV đầu tiên tác động lên mục tiêu phân tử trên tế bào chủ chứ không phải trên virus. Nó được phát triển từ một hợp chất có hoạt tính mạnh nhưng ức chế các kênh ion HERG (Hộp 12.3). Các chất
ức chế kênh ion này thường gây độc hại tính trên tim và do đó, một số lượng lớn chất tương tự đã được tổng hợp để tìm ra một hợp chất ức chế mạnh không chẹn các kênh ion HERG. Maraviroc là kết quả. Đây là một ví dụ về tác nhân hoạt động bằng cách ngăn chặn sự tương tác protein-protein giữa
protein virut và protein tế bào chủ (phần 10.5)
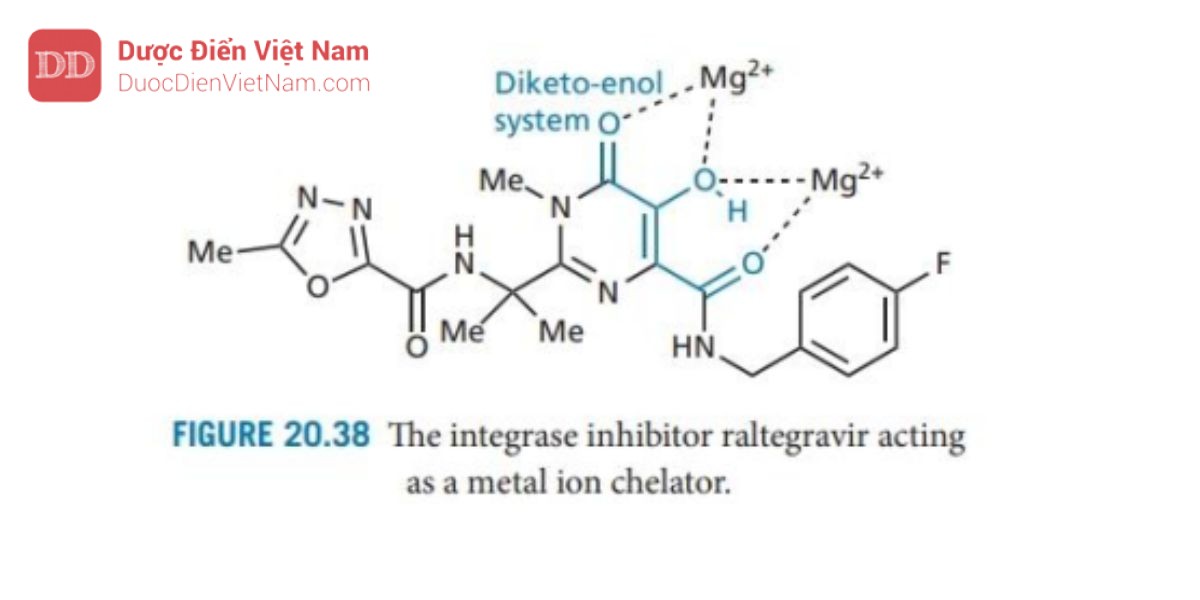
Chất ức chế integrase đầu tiên được tung ra thị trường vào năm 2007 là raltegravir ( Hình 20.38 ). Cân bằng keto-enol rất quan trọng đối với hoạt tính vì nó hoạt động như một nhóm chelat cho hai đồng yếu tố ion magie trong vị trí hoạt động của enzyme.
THUỐC KHÁNG VIRUS ARN: HIV. Tải file PDF tại đây.