




XÁC ĐỊNH DUNG MÔI TỒN DƯ (Phụ lục 10.14) – Dược điển Việt Nam 5
Nếu nội dung bài viết chưa chính xác, vui lòng thông báo cho chúng tôi tại đây
Th8
Quy trình mô tả trong phụ lục này được áp dụng để xác định dung môi tồn dư trong các trường hợp:
1. Định tính dung môi nhóm 1 và dung môi nhóm 2 tồn dư trong dược chất, tá dược, hay dược phẩm;
2. Thử giới hạn dung môi nhóm 1 và dung môi nhóm 2 khi chúng tồn tại trong dược chất, tá dược, hay dược phẩm;
3. Định lượng dung môi nhóm 2 khi lượng tồn dư lớn hơn 1000 phần triệu (0,1 %) hoặc định lượng dung môi nhóm 3 tồn dư khi có yêu cầu.
Các dung môi tồn dư nhóm 1, nhóm 2, nhóm 3 được liệt kê tại các bảng trong Phụ lục 10.14.1 Quy định đối với tạp chất là dung môi tồn dư.
Chuyên luận này giới thiệu 3 cách pha mẫu thử và các điều kiện kỹ thuật tiêm pha hơi các mẫu thử hóa hơi lên hệ thống sắc ký khí. Sử dụng hai hệ sắc ký, hệ sắc ký A thường được chọn trước, còn hệ sắc ký B thường được dùng để củng cố kết quả phát hiện. Việc chọn cách pha mẫu thử tùy thuộc vào độ tan của mẫu thử. Trong một số ít trường hợp cách pha mẫu tùy thuộc dung môi tồn dư cần kiểm tra. Các dung môi tồn dư sau đây không dễ dàng phát hiện được bằng các điều kiện tiêm pha hơi ghi trong chuyên luận này: formamid, 2-ethoxyethanol, 2-methoxyethanol, elhylen glycol, N-methylpyrrolidon và sulfolan. Phải áp dụng các qui trình khác thích hợp để kiểm tra sự tồn dư của các dung môi trên.
Khi dùng qui trình của chuyên luận này để định lượng các dung môi tồn dư, phải tiến hành thẩm định qui trình.
Tiến hành
Thực nghiệm bằng phương pháp sắc ký khí (Phụ lục 5.2) với kỹ thuật tiêm pha hơi tĩnh (static head-space injection).
Pha mẫu thử
Cách 1: Dùng để kiểm tra dung môi tồn dư trong các chất tan trong nước.
Dung dịch mẫu thử (1): Hòa tan 0,200 g mẫu thử trong nước, pha loãng với nước tới 20,0 ml.
Cách 2: Dùng để kiểm tra dung môi tồn dư trong các chất không tan trong nước.
Dung dịch mẫu thử (2): Hòa tan 0,200 g mẫu thử trong N,N-dimethylformamid (DMF) (TT), pha loãng tới 20,0 ml với cùng dung môi.
Cách 3: Dùng để kiểm tra N,N-dimethylacetamid và/hoặc N,N-dimethylformamid khi biết rõ hoặc nghi ngờ có một hoặc cả hai dung môi này tồn dư trong mẫu thử.
Dung dịch mẫu thử (3): Hòa tan 0,200 g mẫu thử trong 1,3-dimethyl-2-imidazolidinon (DMI) và pha loãng đến 20,0 ml với cùng dung môi.
Khi không có cách pha mẫu nào nêu trên phù hợp với mẫu thử, thì cách pha mẫu thử và điều kiện tiêm pha hơi tĩnh áp dụng phải được chứng minh là phù hợp.
Dung dịch dung môi (a): Hòa tan 1,0 ml dung dịch dung môi tồn dư nhóm 1 chuẩn với 9 ml dimethyl sulfoxid (TT) và pha loãng với nước thành 100 ml. Pha loãng 1,0 ml dung dịch này với nước thành 10,0 ml.
Dung dịch dung môi (b): Hòa tan một lượng thích hợp dung môi tồn dư nhóm 2 trong dimethyl sulfoxid (TT). Pha loãng với nước thành 100,0 ml. Tiếp tục pha loãng để thu được dung dịch có nồng độ bằng 1/20 giới hạn quy định tại Bảng 10.14.1-2 trong Phụ lục 10.14.1 Quy định đối với tạp chất là dung môi tồn dư.
Dung dịch dung môi (c): Hòa tan 1,00 g dung môi hoặc các dung môi có trong mẫu thử với dimethyl sulfoxid (TT) hoặc nước (nếu thích hợp), và pha loãng với nước thành 100.0 ml. Tiếp tục pha loãng để thu được dung dịch có nồng độ bằng 1/20 giới hạn quy định tại Bảng 10.14.1-1 hoặc Bảng 10.14.1-2. trong Phụ lục 10.14.1 Qui định đối với tạp chất là dung môi tồn dư.
Dung dịch mẫu trắng: Chuẩn bị như cách pha dung dịch dung môi (c) nhưng không thêm dung môi cần xác định (để kiểm tra sự vắng mặt của các pic nhiễu).
Dung dịch thử: Lấy 5,0 ml dung dịch mẫu thử và 1,0 ml dung dịch mẫu trắng cho vào một lọ đựng mẫu tiêm.
Dung dịch đối chiếu (a) (nhóm 1): Lấy 1,0 ml dung dịch dung môi (a) và 5,0 ml chất pha loãng thích hợp vào một lọ đựng mẫu tiêm.
Dung dịch đối chiếu (a1) (nhóm I): Lấy 1,0 ml dung dịch dung môi (a) và 5,0 ml dung dịch mẫu thử vào một lọ đựng mẫu tiêm.
Dung dịch đối chiếu (b) (nhóm 2): Lấy 1,0 ml dung dịch dung môi (b) và 5,0 ml chất pha loãng thích hợp vào một lọ đựng mẫu tiêm.
Dung dịch đối chiếu (c): Lấy 1,0 ml dung dịch dung môi (c) và 5,0 ml dung dịch mẫu thử vào một lọ đựng mẫu tiêm.
Dung dịch đối chiếu (d): Lấy 1,0 ml dung dịch mẫu trắng và 5,0 ml chất pha loãng thích hợp vào một lọ đựng mẫu tiêm.
Đóng kín các lọ đựng mẫu tiêm nói trên bằng nút cao su có bao lớp polytetratluoroethylen và giữ bởi một vòng chụp ngoài bằng nhôm. Lắc mạnh để có một dung dịch đồng nhất.
Các điều kiện tiêm pha hơi tĩnh có thể dùng:
| Thông số hoạt động | Cách pha mẫu | ||
| 1 | 2 | 3 | |
| Nhiệt độ cân bằng (oC) | 80 | 105 | 80 |
| Thời gian cân bằng (min) | 60 | 45 | 45 |
| Nhiệt độ dòng chảy (oC) | 85 | 110 | 105 |
| Khí mang | Nitrogen hoặc heli dùng cho sắc ký khí ở áp suất thích hợp | ||
| Thời gian điều áp (s) | 30 | 30 | 30 |
| Thể tích tiêm (ml) | 1 | 1 | 1 |
Điều kiện sắc ký
Hệ sắc ký A
Cột mao quản thủy tinh hoặc cột có nòng rỗng, dài 30 m, đường kính trong 0,32 mm hoặc 0,53 mm được phủ bằng lớp các polymer liên kết mạng gồm polydimethylsiloxan 94 % và polycyanopropylphenylsiloxan 6 % (dày 1,8 μm hoặc 3,0 μm).
Khí mang: Nitrogen dùng cho sắc ký khí hoặc heli dùng cho sắc ký khí, tỷ lệ chia dòng 1: 5, tốc độ dòng khoảng 35 cm/s.
Detector ion hóa ngọn lửa [có thể dùng detector khối phổ hoặc để phát hiện dung môi tồn dư nhóm 1 ở dạng hợp chất clor hóa có thể dùng detector thu (bắt) điện tử].
Duy trì nhiệt độ cột ở 40 °C trong 20 min, sau đó gia tăng nhiệt độ với tốc độ 10 °C/min cho đến khi đạt 240 °C, giữ ở 240 °C trong 20 min. Nhiệt độ buồng tiêm 140 °C, nhiệt độ detector 250 °C.
Hệ sắc ký B
Cột mao quản thủy tinh hoặc cột có nòng rỗng, dài 30 m có đường kính trong 0,32 mm hoặc 0,53 mm, được phủ macrogol 20 000 dày 0,25 μm.
Khí mang: Nitrogen dùng cho sắc ký khí hoặc heli dùng cho sắc ký khí, tỷ lệ chia dòng 1 : 5, tốc độ dòng khoảng 35 cm/s.
Detector ion hóa ngọn lửa [có thể dùng detector khối phổ hoặc để phát hiện dung môi tồn dư nhóm 1 ở dạng hợp chất clorid có thể dùng detector thu (bắt) điện tử].
Duy trì nhiệt độ cột ở 50 ° C trong 20 min, sau đó gia tăng nhiệt độ với tốc độ 6 °C /min cho đến khi đạt 165 °C. Giữ ở 165 °C trong 20 min. Nhiệt độ buồng tiêm ở 140 °C, nhiệt độ detector ở 250 °C.
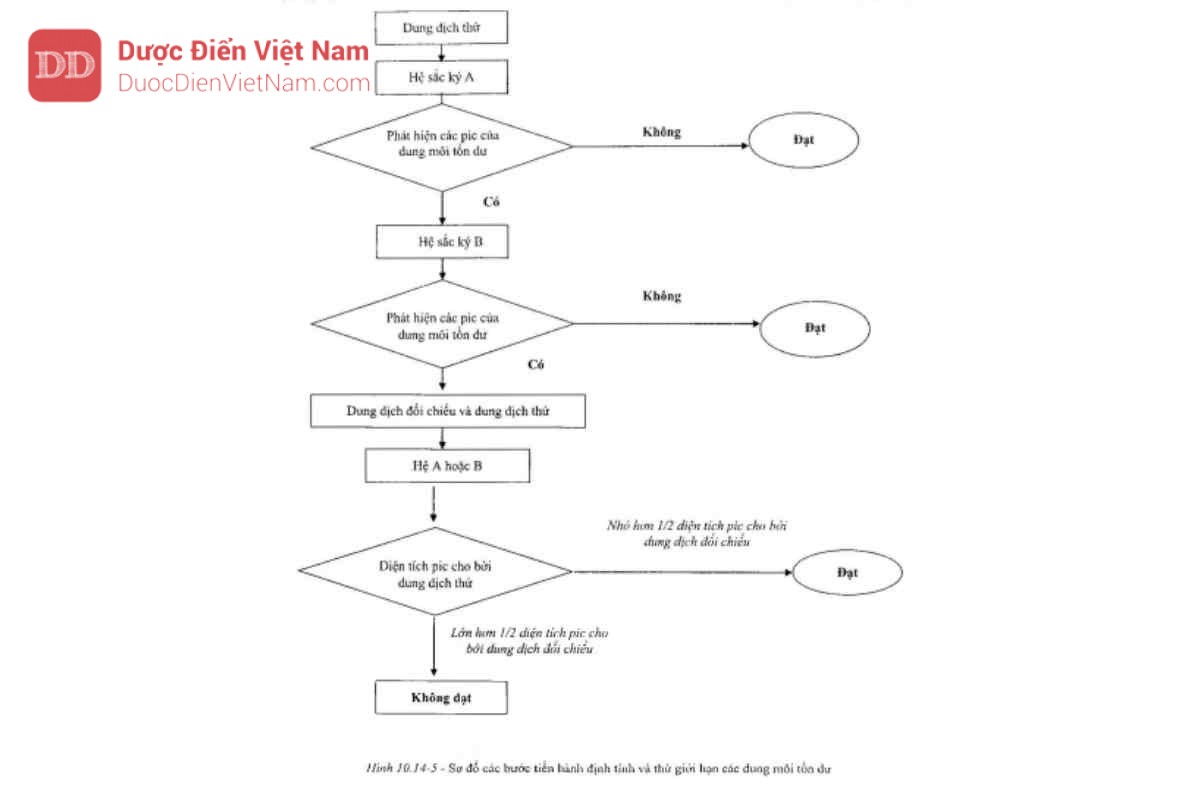
Phân tích trên hệ A
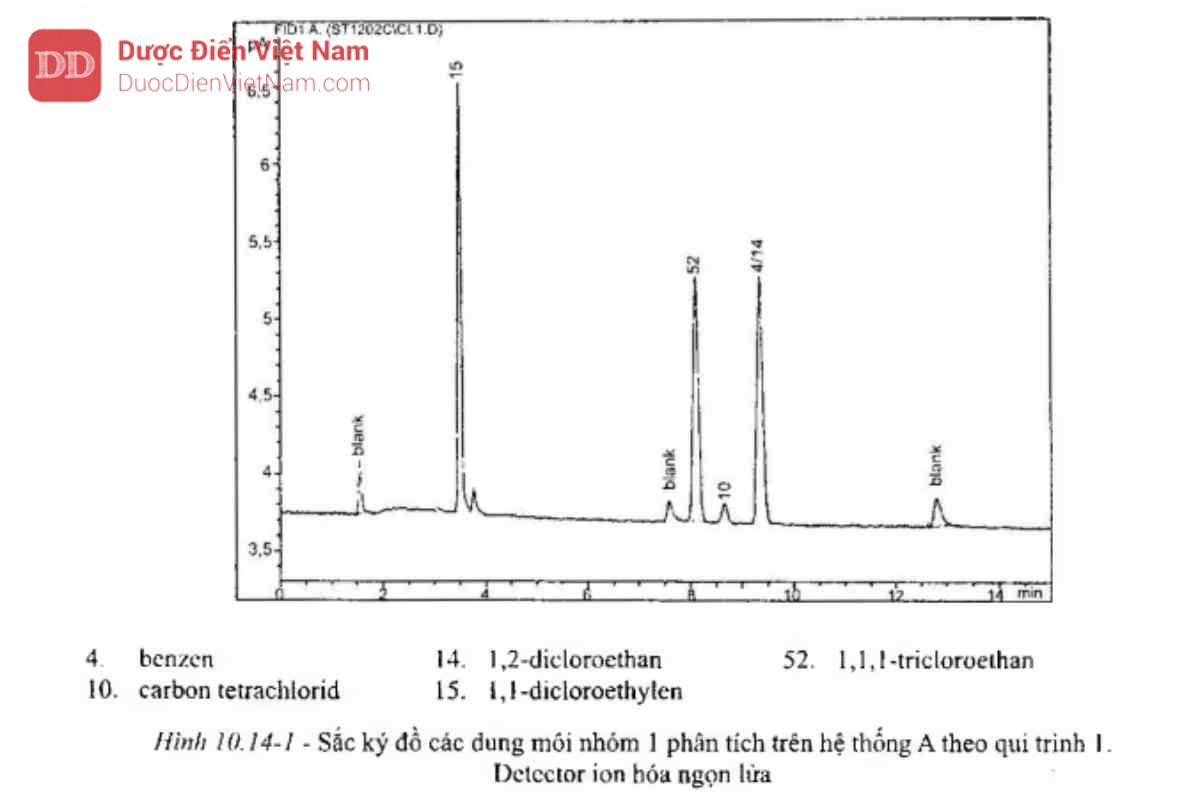
Tiêm 1 ml pha hơi của dung dịch đối chiếu (a), ghi sắc ký đồ ở các điều kiện sao cho có thể xác định được tỷ lệ tín hiệu nhiễu của 1,1,1-tricloroethan. Tỷ lệ tín hiệu/nhiễu của 1,1,1-tricloroethan không được nhỏ hơn 5; xem sắc ký đồ tại Hình 10.14-1.
Tiêm 1 ml pha hơi của dung dịch đối chiếu (a1). Pic của các dung môi nhóm 1 thường được phát hiện.
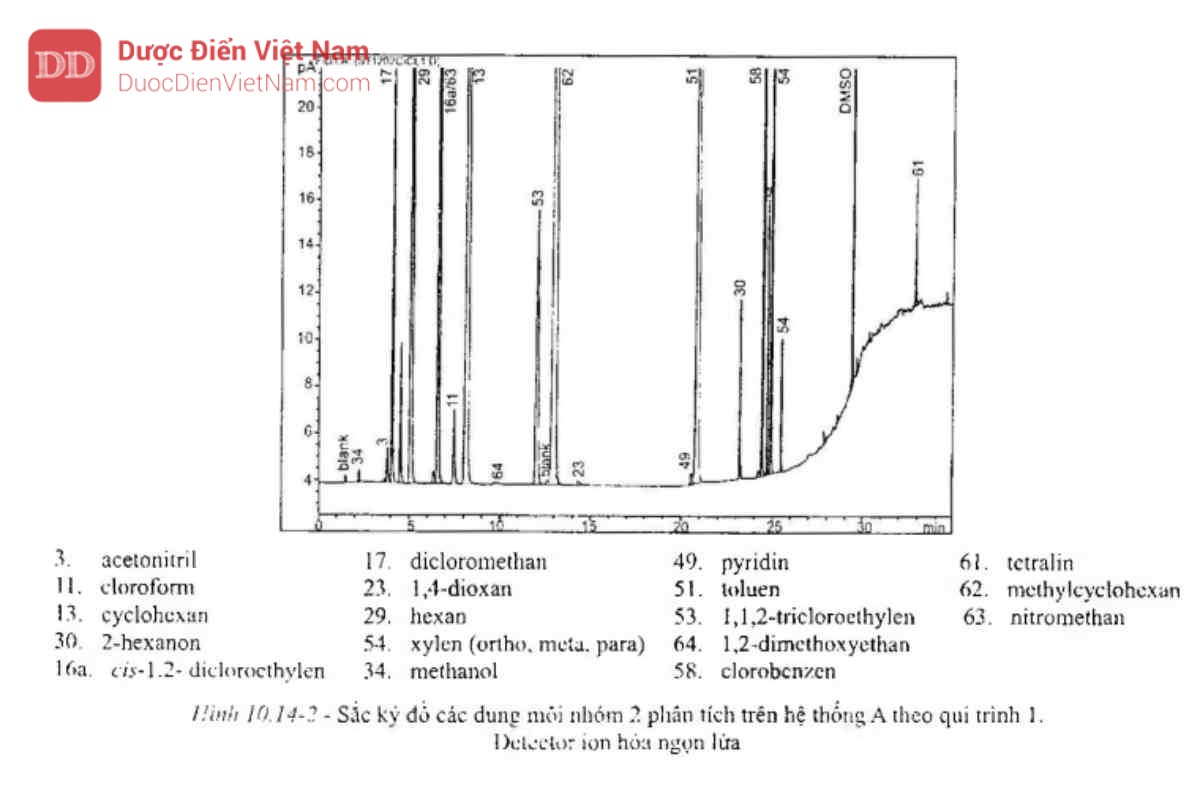
Tiêm 1 ml pha hơi của dung dịch đối chiếu (b), ghi sắc ký đồ ở các điều kiện sao cho hệ số phân giải giữa acetonitril và methylen clorid có thể xác định được. Hệ sắc ký A sẽ thích hợp nếu hệ số phân giải giữa acetonitril và methylen clorid không nhỏ hơn 1,0 và sắc ký đồ có dạng như Hình 10.14-2.
Tiêm 1 ml pha hơi của dung dịch thử lên cột phân tích của hệ A. Nếu sắc ký đồ thu được không có pic nào tương ứng với một trong các pic của những dung môi tồn dư trong sắc ký đồ cho bởi các dung dịch đối chiếu (a) và (b) thì mẫu thử đạt yêu cầu. Nếu sắc ký đồ của mẫu thử có pic tương ứng với pic của một trong những dung môi tồn dư trong sắc ký đồ cho bởi các dung dịch đối chiếu (a) và (b), thì phải tiếp tục phân tích trên hệ B.
Phân tích trên hệ B
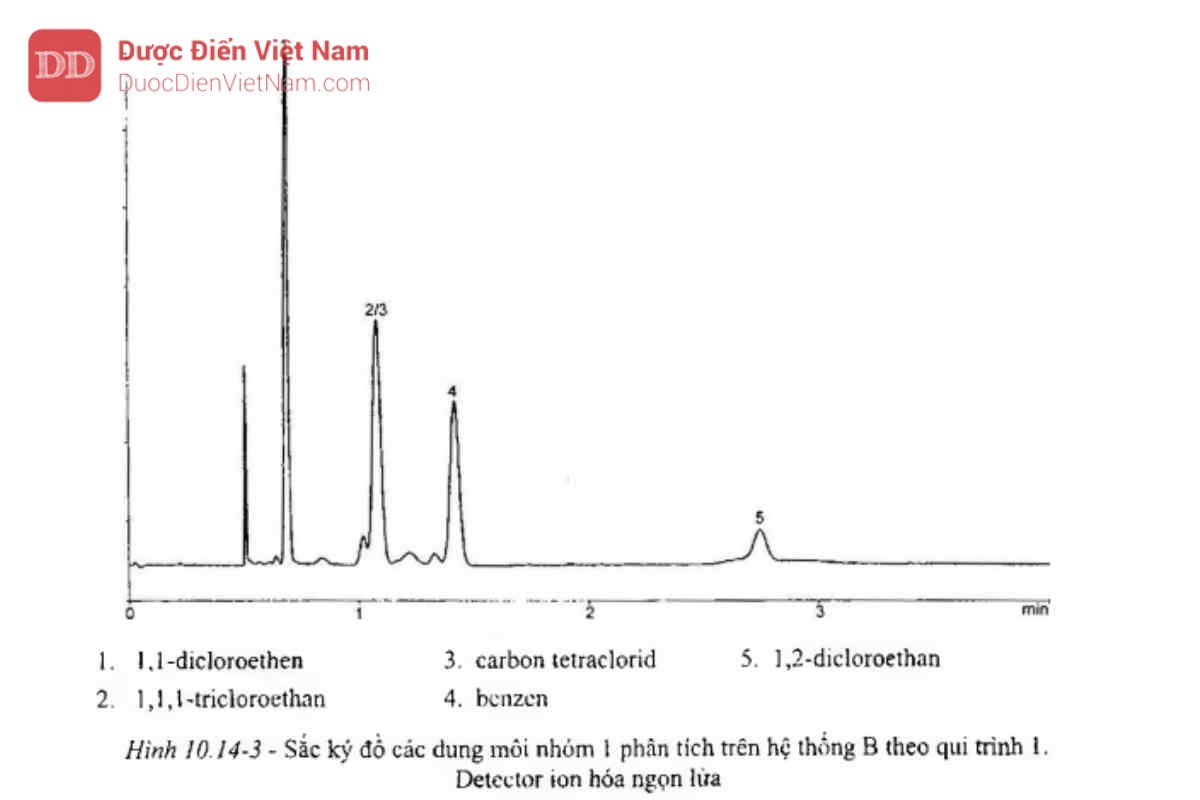
Tiêm 1 ml pha hơi của dung dịch đối chiếu (a) ghi sắc ký đồ ở các điều kiện sao cho có thể xác định được tỷ lệ tín hiệu/nhiễu của benzen. Tỷ lệ tín hiệu/nhiễu của benzen không được nhỏ hơn 5. Xem sắc ký đồ tại Hình 10.14-3.
Tiêm 1 ml pha hơi của dung dịch đối chiếu (a1). Pic của các dung môi nhóm 1 thường được phát hiện.
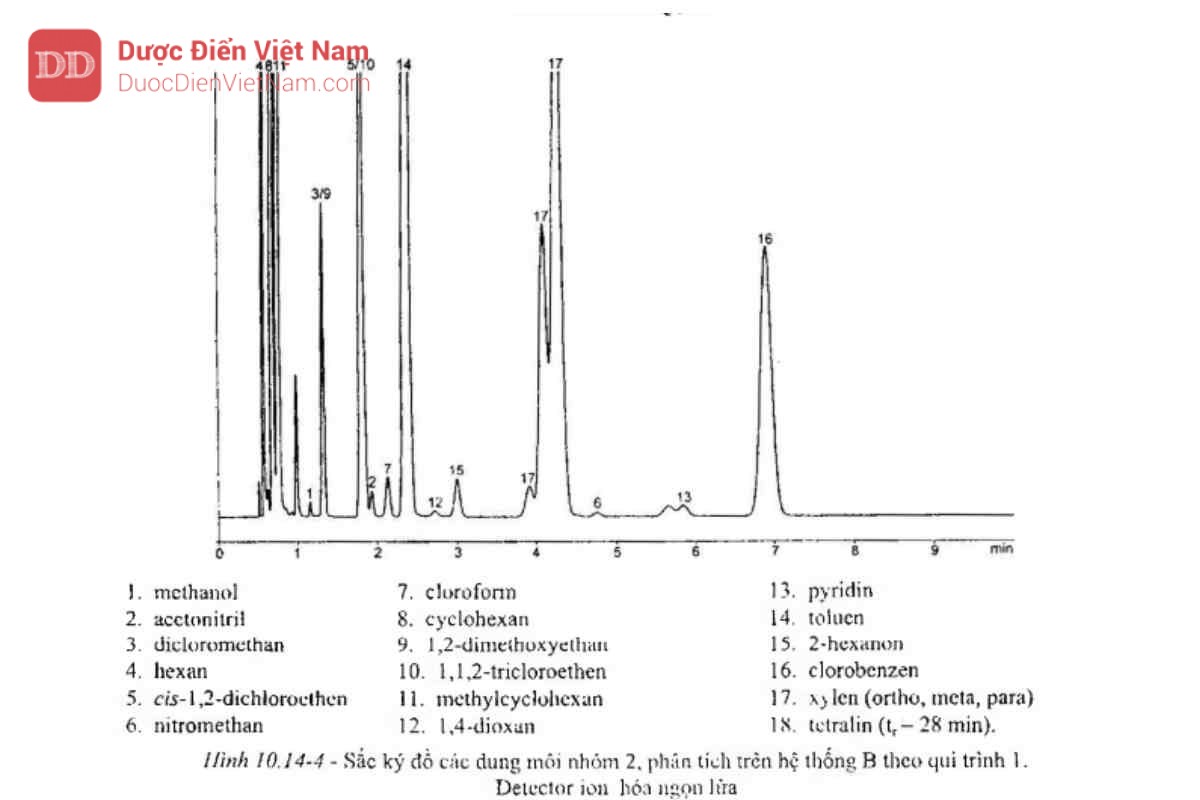
Tiêm 1 ml pha hơi của dung dịch đối chiếu (b), ghi sắc ký đồ ở các điều kiện sao cho hệ số phân giải giữa acetonitril và tricloroethylen có thể xác định được. Hệ sắc ký B sẽ thích hợp nếu hệ số phân giải giữa acetonitril và tricloroethylen không nhỏ hơn 1,0 và sắc ký đồ có dạng như Hình 10.14-4.
Tiêm 1 ml pha hơi của dung dịch thử. Nếu sắc ký đồ thu được không có pic nào tương ứng với một trong các pic của dung môi tồn dư trong sắc ký đồ cho bởi các dung dịch đối chiếu (a) và (b) thì mẫu thử đạt yêu cầu. Nếu sắc ký đồ của mẫu thử có pic tương ứng với một trong những pic của dung môi tồn dư trong sắc ký đồ cho bởi các dung dịch đối chiếu (a) và (b) và phù hợp với kết quả phân tích trên hệ A thì tiến hành như sau:
Tiêm 1 ml pha hơi của dung dịch đối chiếu (c) lên cột phân tích của hệ A hoặc B. Điều chỉnh độ nhạy của hệ thống sao cho chiều cao của pic dung môi tồn dư (hay của các dung môi tồn dư) không dưới 50 % của thang đo.
Tiêm 1 ml pha hơi của dung dịch đối chiếu (d) lên cột. Phải không có pic nhiễu nào được phát hiện. Tiêm 1,0 ml pha hơi của dung dịch thử và 1,0 ml pha hơi của dung dịch đối chiếu (c) lên cột. Tiêm lặp lại 2 lần nữa.
Diện tích pic trung bình của dung môi tồn dư trong sắc ký đồ cho bởi dung dịch thử không được lớn hơn một nửa (1/2) diện tích trung bình của pic tương ứng trong sắc ký đồ cho bởi dung dịch đối chiếu (c). Thử nghiệm chỉ có giá trị nếu độ lệch chuẩn mong đối của các hiệu số giữa ba cặp diện tích pic của chất phân tích thu được từ dung dịch đối chiếu (a) và dung dịch thử nhỏ hơn 15 %. Khi hàm lượng dung môi tồn dư thuộc nhóm 2 hoặc nhóm 3 ở mức 0,1 % hoặc lớn hơn thì có thể tiến hành định lượng bằng phương pháp thêm chuẩn.
10.14.1 Quy định đối với tạp chất là dung môi tồn dư (Theo tài liệu CPMP ICH:283.95)
1. Mở đầu
Mục tiêu của Quy định này là đề ra lượng dung môi cho phép tồn dư trong dược phẩm, nhằm bảo đảm sự an toàn của người bệnh. Quy định khuyến cáo dùng các dung môi ít độc và đưa ra những giới hạn độc tính có thể chấp nhận được đối với một số dung môi.
Dung môi tồn dư trong dược phẩm là các chất hữu cơ bay hơi, được sử dụng hoặc sinh ra trong quá trình sản xuất các dược chất, tả được hoặc trong quá trình bảo chế các dược phẩm. Các dung môi này không loại bỏ được hoàn toàn trong quá trình sản xuất. Sự chọn lọc dung môi thích hợp dùng trong tổng hợp các dược chất có thể nâng cao sản lượng hoặc quyết định các đặc tính như dạng tinh thể, độ tinh khiết, tính hòa tan của sản phẩm. Vì vậy, đôi khi dung môi là một yếu tố quyết định trong quy trình tổng hợp. Quy định này không đề cập đến các dung môi dùng có cần nhắc kỹ lưỡng như một chất phụ gia, hoặc đến các solvat. Tuy nhiên hàm lượng dung môi trong các sản phẩm loại này phải được xác định và chứng minh hợp lý.
Vì các dung môi tồn dư không có tác dụng điều trị, các dung môi này phải được loại bỏ đến mức tối đa để đạt được các yêu cầu kỹ thuật của sản phẩm, việc thực hành tốt sản xuất (GMP) hoặc các yêu cầu chất lượng khác. Dược phẩm phải chứa một mức dung môi tồn dư không được cao hơn các dữ liệu an toàn. Phải tránh dùng một số dung môi có độc tính không thể chấp nhận được (dung môi Nhóm 1, Bảng 10.14.1-1), trừ khi lợi ích của việc sử dụng chúng được xác định chắc chắn. Một số dung môi có độc tính ít nguy hiểm hơn (dung môi Nhóm 2, Bảng 10.14.1-2) cũng cần phải dùng hạn chế, để bảo vệ người bệnh khỏi tác dụng độc hại. Tốt nhất phải dùng các dung môi ít độc (dung môi Nhóm 3, Bảng 10,14.1-3).
2. Phạm vi áp dụng
Các dung môi tồn dư trong dược chất, tá được và dược phẩm thuộc phạm vi áp dụng của quy định này. Vì thế phải thực hiện phép thử tìm các dung môi tồn dư trong quá trình sản xuất hay tinh chế để kiểm soát sự hiện diện của chúng. Chỉ cần kiểm tra đối với các dung môi đã được sử dụng hay được sản sinh ra trong quá trình sản xuất hoặc tinh chế các dược chất, tá dược hoặc được phẩm đó. Mặc dù các nhà sản xuất có thể chọn phương pháp xác định hàm lượng dung môi tồn dư trong sản phẩm, ta vẫn có thể tính hàm lượng đó, đi từ hàm lượng dung môi tồn dư trong các thành phần dùng để sản xuất ra sản phẩm đó. Nếu kết quả tính toán bằng hoặc thấp hơn giới hạn cho phép đã được khuyến cáo trong bản quy định này, thì không cần tiến hành thí nghiệm trên sản phẩm. Trái lại, nếu kết quả tính được vượt mức giới hạn cho phép, dược phẩm phải được thử nghiệm để biết chắc chắn lượng tồn dư dung môi đó có nằm trong phạm vi cho phép không. Sản phẩm cũng phải được thử nghiệm nếu có dùng dung môi trong quá trinh sản xuất.
Quy trình này không áp dụng cho dược chất, tá được hoặc sản phẩm mới đang trong quá trình nghiên cứu áp dụng lâm sàng và cũng không áp dụng cho các dược phẩm đang được lưu hành trên thị trường.
Quy định này áp dụng cho mọi dạng bào chế và mọi cách dùng thuốc. Trong một vài trường hợp, có thể cho phép giới hạn dung môi tồn dư cao hơn như khi dùng trong thời gian ngắn (30 ngày hay ít hơn), hoặc khi dùng dạng thuốc đắp. Việc chứng minh sự đúng dẫn của giới hạn cao này phải dựa trên cơ sở của từng trường hợp một. Xem Chú thích 2 (ở bên dưới) để biết thêm thông tin cơ bản có liên quan tới các dung môi tồn dư
3. Các nguyên tắc cơ bản
Phân loại dung môi tồn dư theo mức độ nguy hiểm
Thuật ngữ “liều có thể dung nạp được cho mỗi ngày” (tolerable daily intake, TDI) được Chương trình quốc tế về an toàn hóa chất (IPCS) sử dụng và thuật ngữ “liều có thể dùng mỗi ngày” (acceptable daily intake ADI) được Tổ chức Y tế Thế giới, các Viện và các cơ quan quản lý sức khỏe quốc gia và quốc tế khác sử dụng để chỉ giới hạn hàm lượng các hóa chất độc có thể dùng mỗi ngày.
Thuật ngữ mới “liều phơi nhiễm được phép mỗi ngày” (permitted daily exposure, PDE) được nêu trong Quy định này là một lượng dung môi tồn dư trong dược phẩm có thể đưa vào cơ thể, để tránh nhầm với liều ADI của cùng dung môi.
Các dung môi tồn dư ghi trong Quy định này được liệt kê theo tên thông thường. Chúng được phân làm 3 nhóm, tùy thuộc khả năng gây độc đối với sức khỏe con người, như sau:
Nhóm I: Các dung môi phải tránh sử dụng
Các chất gây ung thư cho người hay có khả năng gây ung thư cho người rõ rệt. Các chất gây nhiễm độc môi trường.
Nhóm 2: Các dung môi phải hạn chế sử dụng
Các chất gây ung thư trên động vật, không độc cho gen hoặc các tác nhân có thể gây độc không hồi phục như độc tính trên thần kinh hoặc gây quái thai. Các dung môi nghĩ có độc tính quan trọng, nhưng hồi phục được.
Nhóm 3: Các dung môi độc tính thấp
Các dung môi có độc tính thấp trên người: Không cần xác định liều gây tác hại cho sức khỏe.
Các dung môi Nhóm 3 có liều phơi nhiễm được phép mỗi ngày (PDE) bằng hoặc lớn hơn 50 mg/ngày.
Phương pháp xác định giới hạn phơi nhiễm
Phương pháp dùng để xác định liều PDE của các dung môi tồn dư được giới thiệu trong Chú thích 3. Tóm tắt các dữ liệu về độc tính dùng để thiết lập PDE được công bố trong Pharmeuropa Vol 1, No 1, Supplement April/1997.
Phương pháp xác định giới hạn dung môi Nhóm 2
Có hai cách có thể áp dụng để xác định giới hạn cho các dung mỗi Nhóm 2.
Cách 1:
Có thể dùng hàm lượng (nồng độ) giới hạn tính theo phần triệu (ppm) ghi trong Bảng 10.14.1-2. Các nồng độ này được tính theo công thức (1) với lượng chế phẩm dùng mỗi ngày cho là 10 g.
Nồng độ (phần triệu) – (1000 × PDE) /10 (1)
Trong đó:
PDE là liều phơi nhiễm được phép mỗi ngày tính theo mg/ngày;
10 là số lấy làm lượng chế phẩm dùng trong mỗi ngày và tính theo g/ngày.
Giới hạn này áp dụng cho mọi dược chất, tá dược hoặc dược phẩm. Cách 1 có thể áp dụng nếu liều dùng hàng ngày không được biết hoặc không được quy định cụ thể. Khi mọi tá dược, dược chất có trong công thức bào chế đáp ứng với giới hạn cho bởi cách 1, các thành phần của chế phẩm có thể dùng theo bất kỳ tỷ lệ nào. Không cần tính toán gì thêm nếu liều dùng mỗi ngày không quá 10 g. Chế phẩm có liều dùng lớn hơn 10 g mỗi ngày phải tính theo cách 2.
Cách 2:
Không nhất thiết mỗi thành phần của dược phẩm đều phải đáp ứng giới hạn đã đưa ra trong cách 1. Có thể dùng PDE tính theo mg/ngày ghi trong Bảng 10.14.1-2, cùng với liều tối đa mỗi ngày và công thức (1) để xác định hàm lượng dung môi tồn dư cho phép trong dược phẩm. Các giới hạn này được chấp nhận miễn là chứng minh được rằng dung mỗi tồn dư đã được giảm đến lượng thực tế tối thiểu. Các giới hạn này phải thực tế có liên quan đến độ chính xác của phép phân tích, điều kiện sản xuất, các thay đổi hợp lý của quá trình sản xuất và các giới hạn phải thực hiện các tiêu chuẩn công nghệ đương thời.
Có thể áp dụng cách 2 bằng cách cộng các lượng dung môi tồn dư trong mỗi thành phần của chế phẩm. Tổng lượng dung môi dùng trong ngày phải thấp hơn PDE.
Hãy xét một ví dụ áp dụng cách 1 và cách 2 cho dung môi acetonitril trong một dược phẩm. PDE của acetonitril là 4,1 mg/ngày. Như vậy, giới hạn tính theo cách 1 là 410 phần triệu (410 ppm). Lượng dùng tối đa mỗi ngày của dược phẩm là 5,0 g, dược phẩm chứa 2 tá dược. Thành phần dược phẩm và hàm lượng tối đa của acetonitril tồn dư thể hiện trong bảng sau đây:
| Thành phần | Lượng trong công thức bào chế | Hàm lượng acetonitril | Lượng phơi nhiễm mỗi ngày |
| Dược chất | 0,3 g | 800 × 10-6 | 0,24 mg |
| Tá dược 1 | 0,9 g | 400 × 10-6 | 0,36 mg |
| Tá dược 2 | 3,8 g | 800 × 10-6 | 3,04 mg |
| Dược phẩm | 5,0 g | 728 × 10-6 | 3.64 mg |
Tá dược 1 đáp ứng giới hạn tính theo cách 1.
Tá dược 2 dược chất và dược phẩm không đáp ứng giới hạn tính theo cách 1.
Tuy thế, dược phẩm tính theo cách 2 đáp ứng giới hạn 4,1 mg /ngày và như vậy phù hợp với yêu cầu của quy định này.
Hãy xét một ví dụ khác, coi acetonitril là dung môi tồn dư. Lượng dùng tối đa mỗi ngày của một dược phẩm là 5,0 g, dược phẩm có chứa 2 tá được. Thành phần dược phẩm và hàm lượng tối đa của acetonitril tồn dư thể hiện trong bảng sau:
| Thành phần | Lượng trong công thức chế | Hàm lượng acetonitril | Lượng phơi nhiễm mỗi ngày |
| Dược chất | 0,3 g | 800 × 10-6 | 0,24 mg |
| Tá dược 1 | 0,9 g | 2000 × 10-6 | 1,80 mg |
| Tá dược 2 | 3,8 g | 800 × 10-6 | 3,04 mg |
| Dược phẩm | 5,0 g | 1016 × 10-6 | 5,08mg |
Trong ví dụ này, dược phẩm không đáp ứng giới hạn tính theo cách 1 và cả cách 2 bằng cách cộng. Nhà sản xuất phải thử nghiệm dược phẩm để xem quá trình bào chế có làm giảm mức độ tồn dư của acetonitril không. Nếu suốt quá trình bào chế, mức acetonitril không giảm tới giới hạn cho phép, khi đó nhà sản xuất phải tiến hành các bước làm giảm lượng acetonitril trong dược phẩm. Nếu tất cả các bước này không làm giảm được mức tồn dư của acetonitril, trong trường hợp đặc biệt, nhà sản xuất có thể cung cấp bản tóm tắt những cố gắng để giảm lượng dung môi tồn dư, nhằm đạt quy định này, và đưa ra lý lẽ phân tích nguy cơ lợi ích khi dùng chế phẩm có chứa dung môi tồn dư ở mức cao này.
Qui trình phân tích dung môi tồn dư
Phương pháp phân tích dung môi tồn dư thông thường là dùng kỹ thuật sắc ký như sắc ký khí. Nếu thích hợp, dùng phương pháp xác định dung môi tồn dư mô tả trong dược điển. Ngoài ra, nhà sản xuất có thể tự do chọn lựa một quy trình phân tích có hiệu lực thích hợp nhất để áp dụng riêng. Nếu chỉ hiện diện dung môi Nhóm 3 thôi, có thể dùng một phương pháp không đặc hiệu như phương pháp Xác định mất khối lượng do làm khô (Phụ lục 9.6).
Thẩm định các phương pháp xác định dung môi tồn dư phải tuân thủ các quy định của ICH (Hội nghị quốc tế về hài hòa các yêu cầu kỹ thuật để đăng ký các thuốc dùng cho người) ghi trong “Văn bản thẩm định quy trình phân tích” (Text on validation of analytical procedures) và “Mở
rộng văn bản thẩm định quy trình phân tích” (Extension of the ICI Text on validation of analytical procedures).
Báo cáo mức dung mới tồn dư
Nhà sản xuất dược phẩm cần nắm chắc thông tin về nồng độ dung môi tồn dư trong được chất và tá dược để dược phẩm sản xuất ra đạt được mức của Quy định này.
Sau đây là các ví dụ về các thông tin mà nhà cung cấp tá dược hay dược chất có thể cung cấp cho nhà sản xuất dược phẩm. Nhà cung cấp có thể chọn một trường hợp thích hợp trong số các trường hợp sau đây:
Chỉ có các dung môi Nhóm 3 hiện diện. Mất khối lượng do làm khô không quá 0,5 %.
Chỉ có các dung môi Nhóm 2 (X, Y…) hiện diện. Tất cả phải thấp hơn giới hạn tính theo cách 1 (ở đây, nhà cung cấp chỉ rõ tên các dung môi Nhóm 2: X, Y,…).
Chỉ có các dung môi Nhóm 2 (X, Y…) và các dung môi Nhóm 3 hiện diện. Các dung môi Nhóm 2 tồn dư phải dưới mức giới hạn tính theo cách 1 và các dung môi Nhóm 3 tồn dư không quá 0,5%.
Nếu chắc chắn hiện diện các dung môi Nhóm 1 thì phải định tính và định lượng chúng. Dung môi chắc chắn hiện diện là những dung môi được dùng trong giai đoạn cuối của quy trình sản xuất và các dung môi khác được dùng trước đó mà không được loại bỏ hoàn toàn bằng các biện pháp hữu hiệu.
Nếu dung môi Nhóm 2 hoặc Nhóm 3 hiện diện ở mức lớn hơn giới hạn cho phép tính theo Cách 1 (Nhóm 2) hay lớn hơn 0,5 % (Nhóm 3) thì chúng phải được định tính và định lượng.
4. Giới hạn dung môi tồn dư
Dung môi phải tránh sử dụng
Không được sử dụng các dung môi Nhóm I trong việc sản xuất dược chất, tá dược và dược phẩm. Chúng có độc tính không thể chấp nhận được, hoặc có tác hại đến môi trường. Khi buộc phải sử dụng chúng trong quy trình sản xuất một dược phẩm có tác dụng trị liệu vượt trội rõ rệt, phải tuân thủ giới hạn ghi trong Bảng 10.14.1-1; trừ trường hợp buộc phải chấp nhận giới hạn lớn hơn đã được giải thích chứng minh rõ. Dung môi 1,1,1-tricloroethan được xếp trong Bảng 10.14.1-1 vì là một tác nhân nguy hại cho môi trường. Giới hạn 1500 phần triệu dựa trên cơ sở thông tin về dữ liệu an toàn.
Dung môi phải hạn chế sử dụng
Các dung môi trong Bảng 10.14.1-2 phải được quy định mức giới hạn có trong dược phẩm vì độc tính vốn có của chúng. Các giá trị PDE được đưa ra dao động trong khoảng 0,1 mg/ngày. Hàm lượng tồn dư trong dược phẩm dao động trong khoảng 10 phần triệu.
Dung môi có độc tính thấp
Các dung môi Nhóm 3 (có trong Bảng 10.14.1-3) có thể coi như ít độc và có nguy cơ thấp đối với sức khỏe con người. Nhóm 3 bao gồm các dung môi không nguy hiểm đối với sức khỏe con người ở hàm lượng thường được chấp nhận trong dược phẩm. Tuy nhiên chưa có các nghiên cứu về độc tính trường diễn và về khả năng gây ung thư của nhiều dung môi thuộc nhóm này. Các dữ liệu hiện có cho thấy các dung môi này tỏ ra ít độc trong các nghiên cứu cấp diễn hoặc ngắn hạn và cho kết quả âm tính với những thử nghiệm độc tính trên gen. Do đó, coi như được phép dùng các dung môi này với lượng 50 mg mỗi ngày hoặc thấp hơn (tương ứng với hàm lượng 5000 × 106 hoặc 0,5 % tính theo cách 1) mà không cần phải thuyết minh. Có thể dùng ở mức cao hơn, miễn là thực tế các mức đó có liên quan đến khả năng sản xuất và thực hành sản xuất tốt. Dung môi chưa có đủ thông tin về độc tính
Các dung mới trong Bảng 10.14.1-4 có thể cũng được các nhà sản xuất dược chất, tá dược hoặc dược phẩm quan tâm. Tuy nhiên, chưa có các dữ liệu đầy đủ về độc tính của chúng để làm cơ sở cho việc xác định PDE. Khi sử dụng các dung môi này trong sản xuất, nhà sản xuất phải giải trình rõ ràng sự tồn dư của các dung môi này trong sản phẩm của mình.
Bảng 10.14.1-1: Nhóm I, các dung môi phải tránh sử dụng trong dược phẩm
| Dung môi | Nồng độ giới han (μg/g) | Độc tính |
| Benzen | 2 | Gây ung thư |
| Carbon tetraclorid | 4 | Độc và nguy hại cho môi trường |
| 1,2- Dicloroethan | 5 | Độc |
| 1,1- Dicloroethen | 8 | Độc |
| 1,1,1- Tricloroethan | 1500 | Nguy hại cho môi trường |
Bảng 10,14.1-2: Nhóm 2, các dung môi phải hạn chế sử dụng trong dược phẩm
| Dung môi | Liều phơi nhiễm được phép mỗi ngày (PDE) (mg/ngày) | Nồng độ giới hạn (μg/g) |
| Acetonitril | 4,1 | 410 |
| Clorobenzen | 3,6 | 360 |
| Cloroform | 0,6 | 60 |
| Cyclohexan | 38,8 | 3880 |
| 1,2-Dicloroethen | 18,7 | 1870 |
| Dicloromethan | 6,0 | 600 |
| 1,2-Dimethoxyethan | 1,0 | 100 |
| N,N-Dimethylacetamid | 10,9 | 1090 |
| N,N-Dimethylformamid | 8,8 | 880 |
| 1.4- Dioxan | 3,8 | 380 |
| 2- Ethoxyethanol | 1,6 | 160 |
| Ethylenglycol | 6,2 | 620 |
| Formamid | 2,2 | 220 |
| Hexan | 2,9 | 290 |
| Methanol | 30,0 | 3000 |
| 2- Methoxyethanol | 0,5 | 50 |
| Methylbutylketon | 0,5 | 50 |
| Methylcyclohexan | 11,8 | 1180 |
| N-Methylpyrrolidon | 5,3 | 530 |
| Nitromethan | 0,5 | 50 |
| Pyradin | 2,0 | 200 |
| Sulfolan | 1,6 | 160 |
| Tetrahydrofuran | 7,2 | 720 |
| Tetralin | 1,0 | 100 |
| Toluen | 8,9 | 890 |
| 1,1,2- Tricloroethen | 0,8 | 80 |
| Xylen* | 21,7 | 2170 |
* Thường dùng hỗn hợp gồm m-xylen 60%; p-xylen 14%; o-xylen 9% và ethyl benzen 17%.
Bảng 10.14.1-3: Nhóm 3, các dung môi phải được giới hạn vì GMP hoặc vì các yêu cầu chất lượng khác
| Acid acetic | Heptan |
| Aceton | Isobutyl acetat |
| Anisol | Isopropyl acetat |
| 1- Butanol | Methyl acetat |
| 2- Butanol | 3- Methyl-1-butanol |
| Butyl acetat | Methylethylketon |
| Tert-Butylmethyl ether | Methylisobutylketon |
| Cumen | 2- Methyl-1-propanol |
| Dimethylsulfoxid | Pentan |
| Ethanol | 1- Pentanol |
| Ethyl acetat | 1- Propanol |
| Ethyl ether | 2- Propanol |
| Ethyl format | Propyl acetat |
| Acid formic |
Bảng 10.14.1-4: Các dung môi chưa có đủ thông tin về độc tính
| 1,1- Diethoxypropan | Methylisopropylketon |
| 1,1-Dimethoxymethan | Methyltetrahydrofuran |
| 2,2-Dimethoxypropan | Petroleum |
| Isooctan | Acid tricloracetic |
| Isopropyl ether | Acid trifluoroacetic |
Chú thích 1: Thuật ngữ
Genotoxic carcinogens: Tác nhân gây ung thư do tác động lên gen hay chromosom.
LOEL: Viết tắt của từ “lowest-observed effect level” (mức phát hiện gây hại thấp nhất), liều thấp nhất dùng trong một thử nghiệm hay nhóm các thử nghiệm gây sự gia tăng có ý nghĩa sinh học về tần số hoặc về mức độ nghiêm trọng của bất kỳ các tác dụng nào đó trên người hay động vật bị phơi nhiễm.
Modifying factor: Hệ số hiệu chỉnh, là hệ số được chuyên gia độc chất học xác định và áp dụng cho các dữ liệu định lượng sinh học để tính các thông số an toàn cho người.
Neurotoxicity: Nhiễm độc thần kinh
NOEL: Viết tắt của từ “no-observed effect level” (mức không phát hiện gây hại), liều cao nhất của chất đã dùng mà không gây sự gia tăng có ý nghĩa sinh học về tần số hoặc về mức độ nghiêm trọng của bất kỳ các tác dụng nào đủ trên người hay động vật bị phơi nhiễm.
PDE: viết tắt của từ “Permitted daily exposure”
Permitted duily exposure: Lượng cao nhất có thể dùng trong một ngày của dung môi tồn dư trong dược phẩm.
Reversible toxicity: Độc tính phục hồi được, tức là sẽ mất đi sau khi kết thúc phơi nhiễm.
Strongly suspected human carcinogen: Tác nhân nghi ngờ gây ung thư trên người, một chất không có dấu hiệu dịch tễ học là có khả năng gây ung thư nhưng lại cho kết quả dương tính về độc tính trên gen và các dấu hiệu rõ ràng về khả năng gây ung thư trên loài gặm nhấm.
Teratogenicity: Độc tính gây quái thai, sự cố gây dị tật về cấu trúc trong quá trình phát triển thai nhi khi sử dụng một chất trong thời kỳ mang thai.
Chú thích 2:
2.1. Quy chế bảo vệ môi trường đối với các dung môi hữu cơ bay hơi
Một số dung môi thường dùng trong sản xuất dược phẩm được liệt kê như các hóa chất độc trong các chuyên luận EHC (Environmental Health Criteria) và IRIS (Integrated Risk Information System). Trong các mục tiêu của các tổ chức như “Chương trình thế giới về an toàn hóa chất” (IPCS = International Programme on Chemical Safety), “Cơ quan bảo vệ môi trường Mỹ (USEPA = United States Environmental Protection Agency) và “Cơ quan quản lý thực phẩm và dược phẩm Mỹ (USFDA = United States Food and Drug Administration) có việc xác định các mức phơi nhiễm có thể chấp nhận được. Mục đích là bảo vệ sức khỏe con người và giữ gìn sự trong sạch vẹn toàn của môi trường, chống lại các tác dụng có thể có hại của các hóa chất do phơi nhiễm lâu dài tại môi trường. Các phương pháp dùng để xác định mức tối đa phơi nhiễm an toàn thường được căn cứ trên các thử nghiệm dài hạn. Khi không có các dữ liệu từ những thử nghiệm dài hạn, có thể dùng dữ liệu từ các thử nghiệm ngắn hạn với việc cải tiến cách tiếp cận như việc sử dụng các yếu tố hệ số an toàn lớn hơn. Cách tiếp cận nói ở trên đây chủ yếu liên quan tới các phơi nhiễm dài hạn hay suốt đời, đối với các cộng đồng dân cư, trong môi trường xung quanh như không khí, thức ăn, nước uống và các môi trường khác.
2.2. Dung môi tồn dư trong thuốc
Các giới hạn phơi nhiễm trong Quy định này được thiết lập căn cứ vào các phương pháp và các dữ liệu về độc tính mô tả trong các tiêu chuẩn EHC và trong các chuyên luận của EHC và IRIS. Tuy nhiên, một số giả định đặc biệt về dung môi tồn dư trong việc tổng hợp và bào chế các dược phẩm phải được tính đến khi thiết lập các giới hạn phơi nhiễm. Đó là:
1/ Những người bệnh (không phải toàn thể cộng đồng) phải sử dụng thuốc để phòng hoặc chữa bệnh.
2/ Việc giả định người bệnh bị phơi nhiễm suốt đời là không cần thiết đối với đa số các dược phẩm, song có thể thích hợp như một giả thiết nghiên cứu, nhằm giảm nguy cơ đến sức khỏe con người.
3/ Các dung môi tồn dư là những thành phần không thể tránh được trong sản xuất được phẩm và còn thường là một phần của thuốc.
4/ Các dung môi tồn dư phải không được vượt quá các mức độ khuyến cáo, trừ những trường hợp đặc biệt. 5/ Các dữ liệu từ các nghiên cứu độc chất học dùng để xác định các mức chấp nhận được cho các dung môi tồn dư phải được thiết lập bằng cách sử dụng các quy trình thích hợp, chẳng hạn các quy trình do OECD mô tả và sử dụng cuốn sách đỏ của Tổ chức quản lý thực phẩm và dược phẩm của Mỹ (FDA Red book)
Chú thích 3: Phương pháp thiết lập các giới hạn phơi nhiễm Phương pháp đánh giá nguy cơ của Gaylor – Kodell (Xem: Gaylor, D W. và Kodell, R. L. – Thuật toán nội suy tuyến tính dễ đánh giá liều thấp của các chất độc. J. Environ Pathology, 4, 305, 1980) thích hợp với các dung môi gây ung thư thuộc Nhóm 1. Chỉ trong các trường hợp đã có các dữ liệu đáng tin cậy về tính gây ung thư thì phải áp dụng ngoại suy bằng cách dùng các mô hình toán học để thiết lập các giới hạn phơi nhiễm. Các giới hạn phơi nhiễm của các dung môi Nhóm 1 có thể được xác định với việc sử dụng một thông số an toàn lớn (từ 10.000 đến 100.000) đối với “mức không phát hiện gây hại” (NOEL). Việc phát hiện và định lượng các dung môi này phải được thực hiện bằng các kỹ thuật phân tích tiên tiến.
Các mức phơi nhiễm chấp thuận được của các dung môi Nhóm 2 ghi trong quy định này được thiết lập bằng cách tính các giá trị PDE theo các quy trình xác định giới hạn phơi nhiễm của các thuốc (xem Pharmacopeial Forum 11-12/1989) và theo phương pháp đã được Chương trình thế giới về an toàn hóa chất (IPCS) thừa nhận nhằm đánh giả nguy cơ đối với sức khỏe con người do hóa chất (xem Environmental Health Criteria 170, WHO, 1994). Các phương pháp này tương tự như các phương pháp của Cơ quan bảo vệ môi trường Mỹ (USEPA) và của Cơ quan quản lý thực phẩm và dược phẩm Mỹ (USFDA) và các tổ chức khác nữa. Phương pháp được giới thiệu sơ lược ở đây nhằm cung cấp một hiểu biết tốt hơn nguồn gốc của các liều phơi nhiễm được phép mỗi ngày (PDE). Không nhất thiết phải thực hiện các phép tính toán này mà sử dụng ngay các giá trị PDE đã ghi trong các bảng trình bày ở Mục 4 của Quy định này.
Giới hạn PDE được tính từ “mức không phát hiện gây hại” (NOEL) hoặc từ “mức phát hiện gây hại thấp nhất” (LOEL) trong thử nghiệm trên súc vật như sau:
PDE = (NOEL × thể trọng quy ước)/ (F1 x F2 x F3 x F4 x F5)
Trong đó:
Thể trọng quy ước là khối lượng cơ thể người trưởng thành, không phân biệt giới tính, được quy ước để tính ở đây là 50 kg;
F là các hệ số hiệu chỉnh.
PDE thường được tính từ NOEL. Khi không thiết lập được NOEL, có thể dùng LOEL. Các hệ số hiệu chỉnh để ra ở đây để chuyển đổi các dữ liệu thử nghiệm trên súc vật sang cho người, cũng tương tự như các “hệ số không chắc chắn” (Uncertainty factors) dùng trong “Tiêu chuẩn trong sạch của môi trường” (EFIC) (Xem Environmental Health Criteria 170, WHO, Geneva, 1994) và “hệ số hiệu chỉnh” (modifying factors) hay “hệ số an toàn” (safety factors) dùng trong Pharmacopeial Forum. Giả định 100 % phơi nhiễm toàn thân được sử dụng trong tất cả các tính toán, bất kể dùng thuốc theo đường nào.
Các hệ số hiệu chỉnh
F1 là hệ số ngoại suy giữa các loài:
F1 = 2, khi ngoại suy từ thử nghiệm trên chó sang người;
F1 = 2,5, khi ngoại suy từ thử nghiệm trên thỏ sang người
F1 = 3, khi ngoại suy từ thử nghiệm trên khi sang người
F1 = 5, khi ngoại suy từ thử nghiệm trên chuột cống trắng (rat) sang người;
F1 = 10, khi ngoại suy từ thử nghiệm trên các loài động vật khác sang người;
F1=12 khi ngoại suy từ thử nghiệm trên chuột nhất sang người.
Fl tính được khi so sánh tỷ lệ giữa diện tích bề mặt với khối lượng cơ thể của loài vật tham gia thử nghiệm và của người. Diện tích bề mặt (S) được tính như sau:
S=k.M0,67
Trong đó:
M là khối lượng cơ thể (khối lượng cơ thể của sinh vật tham gia thử nghiệm, dùng trong công thức trên, được liệt kê trong Bảng 10.14.2.5);
k = 10
F2 là hệ số chỉ sự khác biệt giữa các cá thể;
F2 = 10, thường dùng cho tất cả các dung môi hữu cơ và dùng thống nhất trong quy định này;
F3 là một hệ số không hằng định, dùng cho các thử nghiệm phơi nhiễm ngắn hạn;
F3 =1, cho các thử nghiệm kéo dài ít nhất bằng 1/2 đời sống của vật thí nghiệm (1 năm đối với loài gặm nhấm hoặc thỏ, 7 năm đối với chó, mèo hoặc khỉ);
F3 =1, cho các thử nghiệm về sinh sản, kéo dài trong suốt thời gian hình thành các cơ quan phủ tạng;
F3 = 2, cho các thử nghiệm kéo dài 6 tháng, trên loài gặm nhấm hoặc 3,5 năm trên các con thú không thuộc loài gặm nhấm;
F3 = 5, cho các thử nghiệm kéo dài 3 tháng, trên loài gặm nhấm, hoặc 2 năm trên các con thú không thuộc loài gặm nhấm;
F3 = 10, cho các thử nghiệm ngắn hạn hơn.
Trong mọi trường hợp, khi thử nghiệm kéo dài trong khoảng thời gian nằm giữa 2 mốc thời gian quy định ở trên, thì ta cho F3 giá trị cao, ứng với mốc thời gian thử nghiệm ngắn. Ví dụ: F3 = 2 trong thử nghiệm kéo dài 9 tháng trên loài gặm nhấm (vì 9 tháng nằm trong mốc 1 năm và 6 tháng, do đó lấy F3 bằng F3 của mốc 6 tháng). F4 là một hệ số áp dụng trong các trường hợp độc tính nghiêm trọng như: Gây ung thư không độc cho gen, độc tính thần kinh, độc tính gây quái thai. Trong các thử nghiệm về độc tính sinh sản, hệ số này được dùng như sau:
F4 = 1, khi gây độc trên bào thai cùng với gây độc trên mẹ;
F4 = 5, khi gây độc trên bào thai, nhưng không gây độc trên mẹ
F4 = 5, khi gây quái thai cùng với gây độc trên mẹ;
F4 = 10, khi gây quái thai trên bào thai, nhưng không gây độc trên mẹ
F5 là một hệ số không hằng định, áp dụng khi không thiết lập được “mức không phát hiện gây hại” (NOEL). Khi chỉ có thể xác định được “mức gây hại thấp nhất” (LOEL), có thể cho F5 giá trị cao, có thể tới F5 = 10, tuỳ thuộc vào mức độ nghiêm trọng của độc tính.
Khối lượng cơ thể (cân nặng) quy ước cho người trưởng thành, không phân biệt giới tính ở đây là 50 kg. Khối lượng này tương đối thấp hơn khối lượng chuẩn mực (60 kg đến 70 kg) thường áp dụng trong các tính toán cùng loại (tính liều dùng thuốc) là nhằm mục đích an toàn.
Một số người bệnh có cân nặng nhẹ hơn 50 kg, những người bệnh này phải được xem xét điều chỉnh bằng cách dùng hệ số an toàn xác định PDE.
Nếu dung môi có trong một công thức dược phẩm chuyên dùng cho Nhi khoa, ta phải điều chỉnh thích hợp, vì khối lượng cơ thể của trẻ thấp.
Ví dụ áp dụng công thức tính PDE:
Hãy xét một thử nghiệm độc tính của acetonitril trên chuột nhắt, thử nghiệm này được tóm tắt trong Pharmacurapa Vol 9, Nol, Supplement April 1997 page S 24, “mức không phát hiện gây hại” (NOEL) là 50,7 mg/kg/ngày. PDE của acetonitril trong thử nghiệm này được tính như sau:
PDE = (50,7mg.kg-1.ngày-1 × 50kg) / (12 x 10 x 5 x 1 x 1)=4,22 mg/ngày
Trong đó:
F1 = 12 là hệ số ngoại suy từ thử nghiệm trên chuột nhắt sang người;
F2 =10 là hệ số khác biệt giữa các cá thể (thống nhất quy định là F2 = 10);
F3 = 5 khi thời gian thử nghiệm chỉ có 13 tuần (> 3 tháng);
F4 = 1 khi không thấy độc tính nghiêm trọng nào;
F5 = 1 khi “mức không phát hiện gây độc hại” (NOEL) được xác định.
Phương trình dùng cho khi lý tưởng PV – nRT được áp dụng để chuyển hàm lượng chất khí trong thử nghiệm dùng phương pháp xông (inhalation studies) từ đơn vị phần triệu thành đơn vị mg/L hay mg/m3.
Hãy xét thử nghiệm độc tính sinh sản trên chuột trắng bằng cách xông khí carbon tetraclorid (khối lượng phân tử 153,84) đã được tóm tắt trong Pharmaeuropa Vol 9. No1. Supplement April 1997, p.S9:
Dùng công thức 1000 L = 1 m3 để suy ra hàm lượng mg/m3
Bảng 10.14.1-5: Các giá trị dùng tính toán trong tài liệu này
| Cân nặng của chuột cống trắng (Rat) | 425 g |
| Cân nặng của chuột cống trắng có thai (Pregnant rat) | 330 g |
| Cân nặng của chuột nhắt (Mouse) | 28g |
| Cân nặng của chuột nhất có thai (Pregnant mouse) | 30 g |
| Cân nặng của chuột lang (Guinea — pig) | 500 g |
| Cân nặng của khỉ Ấn Độ (Rhesus monkey) | 25 kg |
| Cân nặng của thỏ (có thai hoặc không có thai) | 4 kg |
| Cân nặng của chó săn nhỏ (Beagle dog) | 11,5 kg |
| Dung lượng hô hấp của chuột cống trắng (Rat respiratory volume) | 290 L/ngày |
| Dung lượng hô hấp của chuột nhắt | 43 L/ngày |
| Dung lượng hô hấp của thỏ | 1440 L/ngày |
| Dung lượng hô hấp của chuột lang | 4301 Lngày |
| Dung lượng hô hấp của người | 28800 L/ngày |
| Dung lượng hô hấp của chó | 9000 L/ngày |
| Dung lượng hô hấp của khỉ | 1150 L ngày |
| Lượng nước chuột nhất dùng | 5 ml/ngày |
| Lượng nước chuột trắng dùng | 30 ml/ngày |
| Lượng thức ăn chuột trắng dùng | 30 g/ngày |